2 Introduction Part 2
2.1 Abstract
Mycobacterium tuberculosis (Mtb) is a significant cause of infectious death worldwide. This intracellular pathogen uses many mechanisms to subvert the host immune system and lie undetected before causing active disease. This subtle pathogen is uniquely dependent on host lipids for energy, production of host-damaging materiel, and immune subversion. In the face of antibiotic-resistant Mtb strains, researchers have turned toward lipid biology for potential avenues of novel antitubercular therapy. The sphingolipid family has gained attention for its profound roles in determining cell fate, immune signaling, and antimicrobial activity. We review the state of the field regarding sphingolipids as candidate targets of host-directed antitubercular intervention.
2.2 Introduction
Mtb has co-evolved with humans for millennia and thus possesses an astounding capacity to subvert the host antimicrobial defenses, establishing a latent TB infection that can persist for years before progressing to active TB disease1. Among its adaptations is a profound reliance on host lipids. This intracellular pathogen utilizes host lipids as a source of energy and carbon source and is highly sensitive to subtle changes in its host lipidome2,3. Thus, in recent years, researchers have turned toward lipid biology as a potential route of host-directed therapy. A particular lipid family of interest is the sphingolipids. Sphingolipids are dual cell death and inflammation regulators, and they play a crucial role in host defense against pathogenic infections. Thus, studying the host-pathogen interactions and sphingolipid biology may provide new insights into developing effective TB therapies.
In addition to its direct impact on human health, studying Mtb provides unique opportunities to explore and unravel typical human immune function. As a highly adapted pathogen, Mtb has evolved redundant pathways to subvert host immune defenses, making it an excellent teacher and model for understanding human immune function. This chapter will discuss how host sphingolipid biology intersects with Mtb biology and how this niche class of lipids may provide promising routes for new antitubercular drugs.
2.3 Mtb and the innate immune system: a tug of war in the lung
2.3.1 Infection start & the granuloma
The primary TB infection route is inhaling cough aerosols an active TB patient produces. In daily life, humans constantly experience airborne microbes and particulates – every breath will contain fungal, bacterial, and viral particles that reach the lungs4. Many of these particles are filtered out and destroyed in the sinus cavity5, but many will make their way down to the alveolar sacs, the deepest regions of the deepest regions of the lung. The alveoli are surveilled by resident immune cells called alveolar macrophages6. These resident macrophages use damage recognition receptors and pathogen recognition receptors to probe for cells, objects (like silica dust), or invaders decorated with damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs)7.
In most cases, a macrophage will uptake these foreign or effete particles through phagocytosis and destroy them in a specialized cellular organelle called the lysosome. The destruction of would-be pathogens in the lysosome is directly beneficial for clearing an infection. However, the macrophage will also present peptides from the destroyed pathogen to the adaptive immune system, enabling the host to mount a concerted and long-lived immune response. These processes protect most immunocompetent (non-immunocompromised) individuals from infection by environmental pathogens. However, Mtb is adept at avoiding and subverting these processes.
Mycobacterium tuberculosis has spent millennia adapting to the human immune response. Few microbes are as well-suited for aerosol transmission as mycobacteria: recent work has shown that the waxy, hydrophobic composition of the Mtb cell wall confers a high capacity to remain airborne for long periods8,9. Others have shown repeatedly that many, if not most, TB infections are initiated by an individual Mtb bacillus (first shown in guinea pigs housed in TB wards and recently verified by infecting macaques with aerosolized, barcoded Mtb bacilli)10–13.
Within the lung space, Mtb infects alveolar macrophages in a Trojan Horse-style infection: Mtb bacilli allow themselves to be internalized via phagocytosis, initiating a program of immune subversion that, in a successful infection, culminates in the recruitment of more alveolar macrophages as well as other immune cells including monocyte-derived (hematopoietic) macrophages, neutrophils, natural killer cells, T cells, and dendritic cells, and non-immune fibroblasts. As reviewed by Ehlers and Schaible in 2013, the cyclic recruitment of immune cells to the site of Mtb infection has two simultaneous, opposing effects: (1) encapsulating the infection in a fibrous net of fibroblasts and antimicrobial immune cells, which prevents the spread of Mtb bacilli throughout the lung and (2) serving the bacteria fresh macrophages, which serve as host cells and propagate the ongoing infection14,15.
These self-contradictory roles for the granuloma result in restricted bacterial spread and sustained infection – it is thus how a latent TB infection is maintained for potentially years before activation to active TB disease. In 2009, Cardona proposed a model in which the same individual is “reinfected” repeatedly as infected macrophages spontaneously escape from the granuloma and seed infection in a new site16. In support of this, Lin et al. (2014) used longitudinal scanning to show that macaque granulomas are often sterilized and cleared13. However, this is counteracted by the formation of new granulomas in new sites13. In this now-widely accepted model of granuloma containment of Mtb, a patient with a latent TB infection can control infection for long periods but will proceed to active TB disease if the immune regulation within the granuloma fails.
The dissolution of the granuloma structure is marked by widespread necrotic cell death, caseation (tissue degradation with a “cheese-like” appearance17), and eventual cavitation (total collapse, leaving a hole in the lung space) of the granuloma releases the contained bacteria from its restriction and allows them to replicate uncontrolled. At this point, bacteria and necrotic cellular detritus spill into the airway, and the patient’s coughing spreads the released bacteria.
Shortly, we will discuss how lipids influence the interactions between an infecting Mtb bacillus and an individual host cell. First, though, we must note that lipid dysregulation is a hallmark trait of uncontrolled TB disease, even at the organismal level. Histological studies by Hunter et al. in 2007 showed that activation of latent TB disease occurs in tandem with a spillage of lipidated material from ruptured granulomas into the airway18 – a form of lipidated pneumonia in which cholesterols and cholesteryl esters, triacylglycerols, and lactocylceramides accumulate in the caseum17. In many ways, the moment-to-moment stability/integrity of the granuloma reflects the immunocompetence of the host: immunocompromising events such as HIV, chemotherapy, or malnutrition tip the balance in favor of the invading Mycobacterium tuberculosis. The granuloma’s structure is the product of many host-pathogen interactions, the sum of many individual battles between host and microbe. In the unique Mtb infection, many host-pathogen interactions occur via a lipid or membrane interface. We will now discuss the Mtb lifecycle within a host cell: how Mtb subverts the typical immunological processes of the macrophage.
2.3.2 A Trojan Horse in the belly of the beast
Alveolar macrophages surveil the lung space for would-be pathogens, foreign material, or dying cells. To accomplish this, they express an array of receptors that recognize pathogen-associated or damage-associated molecular patterns (PAMPs and DAMPs, respectively). These receptors, or pathogen-recognition receptors (PRRs), bind to a cognate antigen and initiate a signaling cascade that can trigger phagocytosis, cytokine secretion, and adaptive immune system activation.
As is the case with most bacterial pathogens, a variety of Mtb surface structures are recognized by phagocytic receptors such as Dectin-1, the Mannose Receptor, and DC-SIGN, as well as non-phagocytic receptors such as TLR2. Upon recognizing the Mtb pathogen-particle, the alveolar macrophage internalizes the bacterial cell via phagocytosis. However, unlike other pathogens, Mtb appears to selectively engage receptors that fail to initiate immune signaling. Van Kooyk and others have reported that Mtb selectively binds receptors that fail to initiate inflammatory signaling19,20.
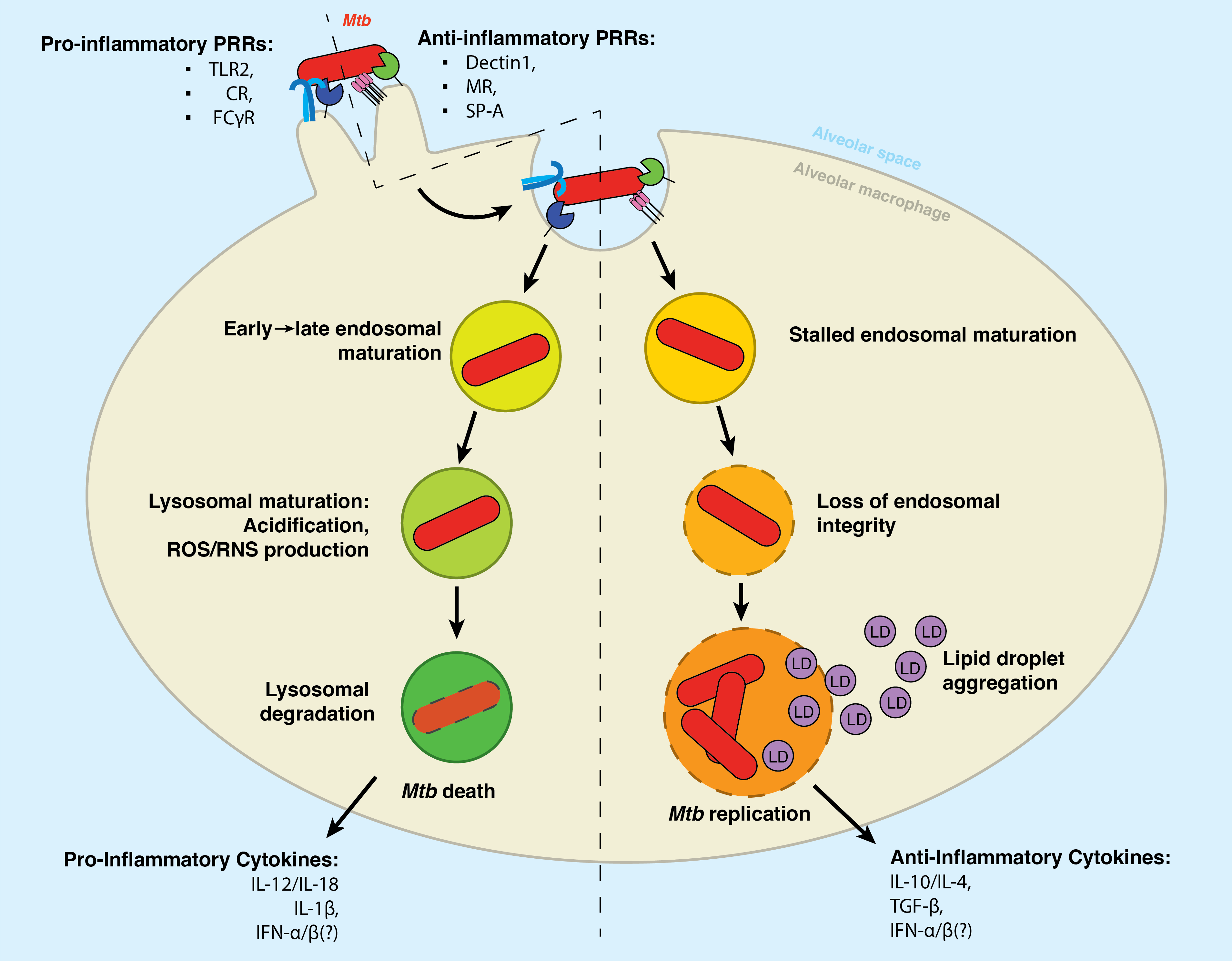
Therein lies the Trojan Horse infection: the macrophage internalizes the Mtb bacillus as a part of its typical antimicrobial program – but Mtb subverts this programming to hijack the cell and enable its survival therein. Much evidence has shown that the polarization of the infected macrophage has an immense impact on whether it will destroy the bacteria or succumb to its machinations21. In 2022, Arish and Naz described that the plasticity of the macrophage might be a therapeutic target for TB therapy: inducing a so-called M1 inflammatory polarization enhances the cell’s capacity to mount an antimicrobial defense against Mtb, among which is the upregulated expression of inflammatory pathogen recognition receptors such as TLR222.
2.3.3 Arresting development: the blockade of lysosomal maturation.
For most phagocytosed particles, the lysosome is the end of the line. This organelle is responsible for destroying unwanted material in the cell. It thus contains hydrolases (which can cleave proteins, lipids, sugars, and nucleic acids), vATPases (which produce a highly acidic pH), and antimicrobial peptides (which are directly bactericidal)7,23.
The contents of the phagosome are delivered to the lysosomal compartment through a series of fusion/fission events with the endosomal compartment. As depicted in the left hand of Figure 2.1, the phagosome will fuse with the early endosome, the late endosome, and then the lysosome. We and others have written extensively on the maturation of the phagosome, and we recommend further reading for an in-depth discussion on standard endosomal trafficking (Chapter 8)24–27.
However, Mtb initiates a cascade of immune subversion once inside the host cell1. Environmental cues such as reduced pH and increased sodium concentration trigger these pathogenic programs, which block the typical maturation of the phagosome and allow the bacteria to survive within an atypical endosomal compartment commonly called the mycobacteria-containing vacuole28. We recommend the 2019 review by Carranza et al. for an in-depth description of the mechanisms that Mtb utilizes to halt phagosomal maturation29.
Following phago/lysosomal blockade, the Mtb-containing vacuole retains many canonical features of the early phagosome30,31. This compartment has a nearly neutral pH, is decorated with early endosomal markers such as Rab530. Additionally, the Mtb-containing phagosome retains Rab14 and Rab22a. Unlike the early endosome, however, the Mtb-contaiing vacuole is largely devoid of Vps34, a class III phosphotidylinositol-3 kinase which produces phosphatidylinositol-3-phosphate (PI(3)P)31. PI(3)P is an essential inducer of autophagy and lysosomal trafficking, and, accordingly, several crucial PI(3)P-binding proteins are not recruited to the Mtb-containing vacuole – including the hepatocyte growth factor-regulated tyrosine kinase substrate (HGS/HRS), which responsible for recruiting the ESCRT complex to initiate lysosomal degradation31,32. Intriguingly, in 2017, Schnettger et al. found that Mtb subverts the Rab20-dependent membrane trafficking pathway to reside in so-called “tight” phagosomes (as opposed to “spacious” phagosomes, which are associated with IFN-γ-induced immunoproteolysis)33.
There are several Mtb pathogenicity factors critical for the subversion of lysosomal maturation. Perhaps the most significant is the Mtb ESX Type VII secretion system34. This secretion system mediates the transfer of many Mtb factors into the lumen of the lysosome, including proteins, lipids, and small molecules35–37. Several of these factors appear to independently serve as membrane-damaging agents – these include the highly characterized ESAT-6/CFP-10 protein heterodimer and the glycolipid phthiocerol dimycocerosate38,39. Other factors directly remodel the phagosomal environment; for example, the terpene nucleoside 1-tuberculosinyladenosine is reported to act as an “antacid” that neutralizes the acid pH in the nascent phagosome40 and eventually induces the formation of lipid droplets41. We will discuss lipid droplets further below.
As will be discussed below, however, this vacuole is dynamic and undergoes dramatic changes as the Mtb continues to overcome the macrophage’s defenses and wrests control over the cell. Prior to phagosomal escape, however, Russell proposed in 2016 a model in which intracellular Mtb will spend an outsize proportion of its time within the endosomal compartment, surreptitiously avoiding immune recognition by existing in a semi-quiescent state42. Among many pieces of supporting evidence, the long-observed paucibacillary quality of the TB lung aligns well with Russell’s model – there are simply very few individual Mtb bacilli in a latent TB infection. Expanding on this model, Russell suggests that it is upon progression to active disease via immune suppression, malnutrition, or some other trigger that the Mtb bacillus will begin a program of phago/endosomal damage42 – a topic discussed further below.
2.3.4 Breaking free: damaging the phagosomal membrane
2.3.4.1 Mtb and other intracellular bacterial pathogens benefit from endo/lysosomal membrane damage
Many intracellular bacterial pathogens utilize phago/lysosomal membrane damage as a mechanism of immune escape; these include Mtb, Salmonella enterica serovar Typhimurium, Lysteria monocytogenes and many others43,44.
The reason for this convergent mechanism of pathogenicity is apparent: by permeabilizing the phagosomal membrane as it matures through the endosomal pathway, the pathogen can inhibit the acquisition of antimicrobial factors45. Chief among these factors is the intraluminal pH of the maturing phagosome: the membrane-bound proton pump vATPase is delivered via fusion with the early endosome shortly after bacterial lumen, which accumulates on the maturing phagosome and acidifies the phagosomal interior23. A variety of pH-regulated antimicrobial factors are simultaneously delivered to the maturing phagosome via fusion with the early endosome, then the late endosome, and finally the lysosome; these include hydrolases which cleave proteins, lipids, and glycans (proteases, lipases, and glycosidases, respectively), as well as pore-forming antimicrobial peptides, and reactive oxygen and nitrogen species46. By perforating the phagosomal membrane, intracellular pathogens such as Mtb prevent the accumulation of these factors within the pathogen-containing vacuole, enabling escape from lysosomal degradation and the presentation of antigens to the adaptive immune system46,47.
To achieve phagosomal perforation, Mtb utilizes a cohort of Type VII secretion systems to transfer soluble pathogenicity factors into the phagosomal lumen – chiefly ESX-1 and ESX-548. Among the most well-characterized of these soluble effectors are EsxA and EsxB – more commonly referred to as early secreted antigenic target-6 (ESAT-6) and culture filtrate protein-10 (CFP-10)48. These proteins are the immunodominant antigens of an Mtb infection and are major determinants of Mtb virulence44,49. Many reports have shown that the loss of either ESAT-6 or CFP-10 results in the significant attenuation of pathogenic Mtb strains49–51. Though not the first to show this, Smith et al. elegantly demonstrated in 2007 that ESAT-6 directly induces the formation of pores in the phago/lysosomal membrane50. Consequently, the permeabilization of the phago/lysosome is a significant driver of Mtb-induced cell death and pathogenicity52.
However, as with other stages of an Mtb infection, membrane damage represents a fulcrum of control for the host cell: the cell has several layers of redundant membrane repair mechanisms which seek to counteract the membranolytic activities of Mtb and regain control over the infection.
2.3.4.2 An arms race for control: Host response to membrane damage Mtb’s inhibition of membrane repair
As described above, the lysosome encloses a toxic milieu of acidic pH, antimicrobial peptides, proteases, lipases, glycosidases, and nucleases53. Independent of pathogenic action, unmitigated lysosomal damage is a potent inducer of cell death54 – thus, the cell utilizes independent and redundant mechanisms of membrane repair. These repair mechanisms operate at different levels of damage and timespan: from the minor and acute to the major and chronic.
In the case of minor membrane damage resulting from an acute membranolytic injury, a suite of proteins called the Sequestosome-like receptors (SLRs), which includes Sequestosome, NDP52, and the galectins55. These SLRs are also used to mark defunct organelles, depolarized mitochondria, protein aggregates, and cytosolic bacteria. These membrane sensors initiate membrane repair by directing the polymerization of components of the ESCRT-III complex. ESCRT-III is composed of a cohort of charged multivesicular body proteins: CHMP1, CHMP2A, -2B, -3, -4A, -4B, -4C and -656. These monomers insert into damaged membranes and polymerize into a spiral which induces membrane curvature around the site of membrane damage. ATP hydrolysis by VPS4 provides the requisite energy to constrict the polymerized ESCRT-III complex and pinch off the targeted membrane region56. This membrane-repairing behavior of the ESCRT-III complex is somewhat ancillary to its primary role of producing multivesicular bodies (MVB) during secretory trafficking: the same complex assembles at the surface of these complex organelles and mediates the scission of microvesicles into the lumen of the MVB56.
In circumstances of major or chronic damage – instances where, despite the efforts of the ESCRT-III complex, an organelle is beyond repair – the deposition of ubiquitin and Sequestosome-like receptors over time induces the recruitment of autophagy machinery56,57. Autophagy results in the destruction of damaged endosome; this is a process used by the cell to degrade defunct or unneeded organelles58. Autophagy involves the construction of a double membrane around a targeted organelle and fusion with to pre-formed lysosomes in order to degrade targeted organelle58. This process is highly dependent on the recruitment of the acyl-CoA synthetase Faa159. This enzyme enables the on-site assembly of phospholipids in the double membrane of the autophagosome, and is critical for the encapsulation of the targeted organelle59.
Autophagy is a central mechanism for restricting Mtb infection60–62. This process that enables the destruction of chronically damaged membranes further enables the proteasomal degradation of the bacterial contents within: the resulting microbe-derived peptides can thus be loaded onto the MHC-II complex and presented to the adaptive immune system via the cross-presentation pathway58. Accordingly, as seen at many other points of the host-pathogen interface, Mtb proactively interferes with the autophagic membrane damage repair response of the host63. In one notable example, Divangazi et al. showed in 2009 that Mtb infection induces membrane damage both at the plasma membrane and the in lysosome, and that the bacteria produces a molecule called lipoxin A4 (LXA4), which blocks prostaglandin E2 (PGE2) synthesis64. These authors show that PGE2 is essential for the recruitment/activation of synaptotagmin, a cytosolic Ca2+ sensor64. Similarly, Mittal et al. showed in 2018 that the secreted Mtb effectors EsxG and EsxH65 directly interfere with the ESCRT response pathway. And as a final example (though this is not to say that these examples are exhaustive), authors such as Saini et al. (and many others) have shown that Mtb suppresses the autophagy response of the cell and directly benefits from the subsequent escape from the adaptive immune response63,66,67.
These examples of the membrane damage response highlight the tit-for-tat manner of an Mtb infection – an arms race over the millennia. As alluded above and as will be discussed further below, the cell’s capacity to respond to and limit the damage caused by Mtb is highly influenced by the immunological priming of the cell prior to infection: a cell primed for antimicrobial effect via IFN-γ stimulation has a higher propensity for maintaining phago/lysosomal integrity and directing the bacterial to lysosomal degradation. It is in an inadequate immune environment that the bacteria is able to inflict major damage to the maturing phagosome and gain cytosolic access.
2.3.5 Lipid droplet formation in late-stage infection
Lipid droplets (LDs) are organelles consisting of a phospholipid monolayer encapsulating a hydrophobic core composed mainly of triacylglycerol species (TG) and sterol esters68. Research suggests that LDs are a central feature of mycobacterial infection, where mature granulomas are replete with “foamy” macrophages69. It is not unique to mycobacteria as many bacteria, viruses, eukaryotic parasites, and fungi are known to induce LDs in infected hosts. Prior reports suggest that LDs are trafficked toward the mycobacteria-containing vacuole in a cytoskeleton-dependent manner70. Further, Barisch et al. showed that Mtb hydrolyzes TG from LDs to fuel its metabolism as a primary source of energy69. Thus, LDs are believed to play a crucial role in the host-pathogen interaction during Mtb infection. Recent work by Bedard et al. showed that individual Mtb pathogenicity factors, such as 1-tuberculosinyladenosine, are sufficient to induce the accumulation of lipid droplets41.
Further research is required to fully understand the mechanisms involved in the formation and utilization of LDs by Mtb, which could lead to developing new therapeutic interventions for TB. Many points in this pathway may serve as targets of antitubercular therapy, including blocking the formation of these organelles, halting the trafficking of LDs to the Mtb-containing vacuole, inhibiting the Mtb phospholipases that hydrolyze TG, or disrupting the Mtb fatty acid β-oxidation pathway. Much research is needed to determine which, if any, of these lipid droplet-targeted therapies may have the most significant effect.
2.3.6 Cytokine production in the granuloma
The cytokines within the granuloma microenvironment hold immense sway in the direction a TB infection can go. Inflammatory cytokines such as Type II interferon (IFN-γ) can potently stimulate the antimicrobial capacity of macrophages, stimulating lysosomal activity, antigen cross-presentation, and heightened production of other inflammatory cytokines such as IL-1β, which further initiate a broad antimicrobial response6. This immune cross-talk allows a fully immunocompetent host to limit the spread of Mtb within the lung and limit bacterial growth6. However, Mtb is also adept at subverting these immune processes – reports by Fortes et al. and others demonstrate that Mtb can prevent the production of inflammatory cytokines and instead induce the secretion of anti-inflammatory cytokines such as TGF-β and suppress the secretion of IFN-γ71,72. Consequently, the overarching immunocompetence of the host can result in feedback loops of either Mtb restriction or Mtb success.
With these feedback loops in mind, cytokine therapy is under study as a potential form of host-directed therapy. Berns et al. reviewed the clinical efficiency of exogenous IFN-γ therapy and found repeated benefits to aerosolized or subcutaneous IFN-γ adjunctive treatment in clinical trials73. Berns et al. write that IFN-γ strongly stimulates antigen presentation and can overcome Mtb’s inhibition of immune processing, resulting in a coordinated and multifaceted attack on the infecting Mtb73. In addition, IFN-γ robustly activates macrophages and stimulates chemotactic migration toward the site of infection. Highly activated macrophages release nitric oxide into the granuloma microenvironment, placing extreme oxidative stress on infected cells and bacteria.
However, some studies have suggested that cytokine therapy can inadvertently cause increased pathology by increasing damage to surrounding lung tissue and exacerbating lung infection74,75. Thus, cytokine therapies, including IFN-γ, are still controversial and subjects of clinical trials. Nonetheless, IFN-γ is a highly cost-effective adjunctive therapeutic in the Russian Federation73. It reduces treatment time and antibiotics, thereby reducing costs and increasing supplies.
2.4 Mtb and Lipids: A recipe for pathogenicity
Many papers describe the dependence of Mtb on host lipid biology. As Ghazaei beautifully wrote in their 2018 review on lipid metabolism in Mtb infection: “If the metabolism of the pathogen is studied, the use of lipid metabolism has been documented”76. In summary, Mtb is an intracellular pathogen with unique lipid dependency, and the full description of this statement would encompass too many topics for this review. However, we will briefly discuss a few examples of Mtb’s unique lipid biology.
A critical phenomenon preceding the progression to active TB disease is the accumulation of “foamy” macrophages in the granuloma77. The accumulation of lipids within these cells gives them their name: these cells have undergone significant dyslipidemia, driving the formation of many lipid droplets within them77. When these cells undergo necroptosis, they release this lipidated material into the granuloma/alveolar space – foamy macrophages are the source of the lipid pneumonia associated with active TB disease. DAG and TAG species have been previously observed to increase upon infection with Mtb78 and are a significant component of TB-associated lipid droplets77. More than a visual cue of metabolic dysfunction in the cell, lipid droplets are a significant energy and carbon source for Mtb69. In some agreement with this, Mtb enzyme Rv2252 is a verified diacylglycerol kinase involved in the production of phosphatidyl-myo-inositol mannosides, Mtb-specific lipids79.
In fact, Mtb depends on host lipids as a primary carbon source during infection. Jain et al. show that Mtb prefers β-oxidation of fatty acids over glycolysis during infection, shifting its metabolism upon entry into the host cell80. These authors found an increase in membrane content of odd-chain lipids in Mtb isolated from infected cells, suggesting that the bacteria selectively shifts to β-oxidation during infection80. Along the same lines, Chandra et al. used inhibitors of fatty acid β-oxidation and found that they severely restrict the intracellular growth of Mtb in tissue culture81, and Yang et al. showed that inhibiting β-oxidation enhances Mtb clearance in a mouse infection model82. Beyond β-oxidation, many reports have found links between Mtb and host cholesterol. Mendum et al. used a transposon mutant library to identify genes that enable Mtb fitness during infection34. Among their findings, they identify a preponderance of cholesterol-catabolizing enzymes that are required for bacterial persistence/growth34. These findings are supported by many similar reports dating back decades83–88.
These are only a few of the many examples of the manners in which lipids are essential for Mycobacterium tuberculosis during infection. We must now shift course briefly to introduce sphingolipids, as this enigmatic class of lipids appears to play decisive roles in Mtb’s struggle for control of the macrophage.
2.5 Sphingolipids overview
2.5.1 Sphingolipid synthesis and trafficking
Sphingolipids are a diverse class of membrane lipids distinguished by a shared sphingoid base. Through much concerted effort, these enigmatic lipids are slowly being recognized for their many roles in neuronal health, development, apoptosis, membrane stability, and signaling capacity. We and others have extensively described the mechanisms of sphingolipid synthesis and trafficking. We recommend the following references for more in-depth reviews of these topics: Chapter 789, Chapter 390, Chapter 4, Hannun, 201891, Wattenberg, 201892, Quinville et al., 202193, and Gault et al., 201094. For this review, we, too, must dive into the sphingolipid synthesis and salvage pathways to begin discussing how sphingolipid biology may influence a Mycobacterium tuberculosis infection.
As depicted in Figure 2.2, sphingolipid production begins in the endoplasmic reticulum. The first and rate-limiting step of sphingolipid synthesis is the condensation of the amino acid L-serine with palmitoyl-CoA via the serine palmitoyltransferase complex (SPT) to produce the transient product 3-ketodihydrosphingosine (KDS). SPT is composed of two primary subunits (SPTLC1 and SPTLC2, though SPTLC3 appears partially redundant with SPTLC2) and acts in tight conjunction with two downstream enzymes: 3-ketodihydrosphingosine reductase reduces KDS to dihydrosphingosine and a cohort of [dihydro]ceramide synthases (CerS1-6) append a secondary acyl chain of varying length. Dihydroceramide Δ4-desaturase (DES) reduces dihydroceramide to produce ceramide (Cer), the nexus of sphingolipid metabolism. After all these initial steps of sphingolipid synthesis, it is from Cer that all complex sphingolipids will be made.
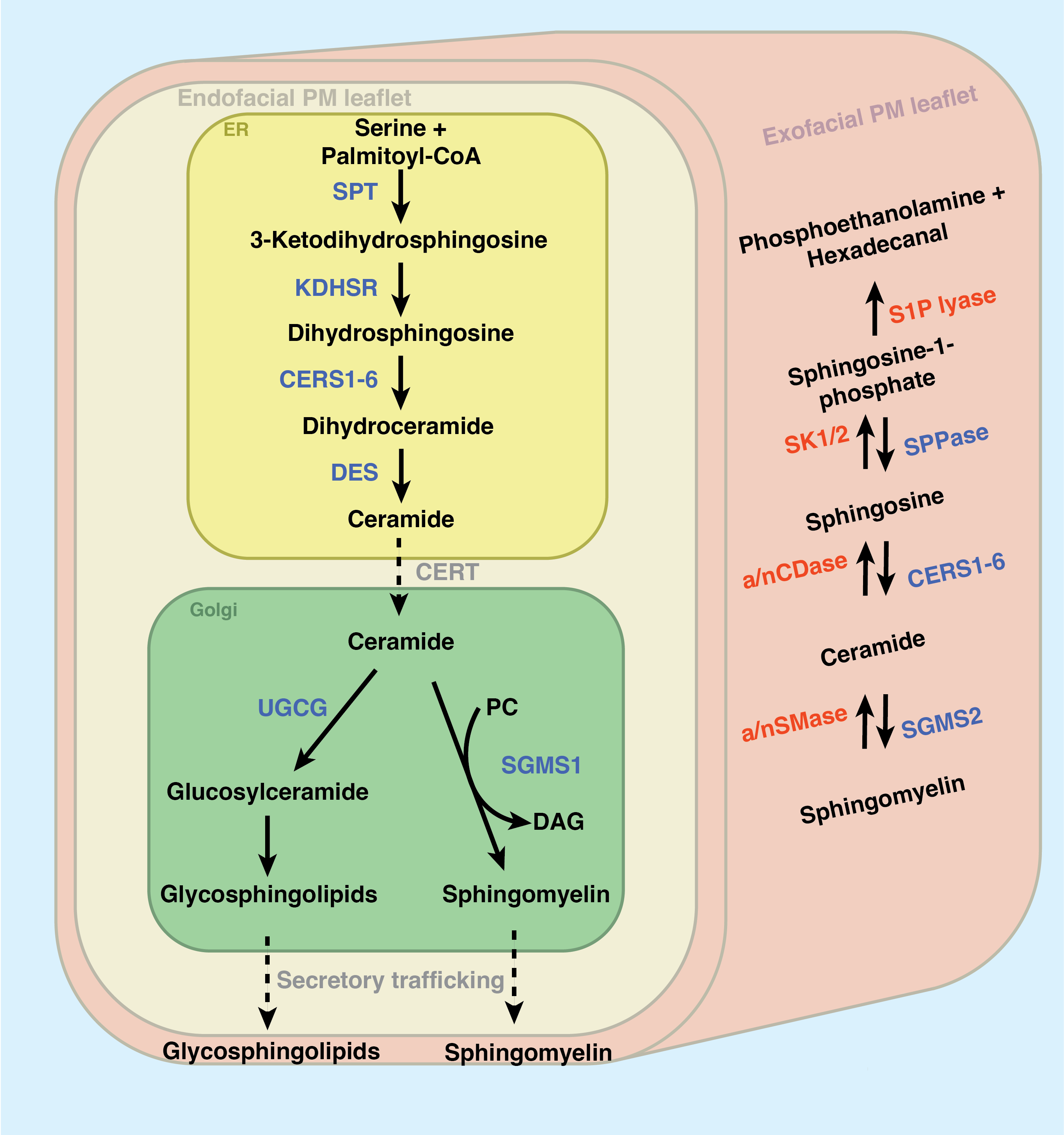
Why maintain the redundancy of six individual ceramide synthases? Recent work has shown that CerS enzymes prefer appending fatty acyl chains of distinct lengths (with some overlap), and different tissues express different proportions of each enzyme. CerS selectivity for chain length was excellently reviewed by Ho et al. in 202295 – here, briefly: CerS1 is most expressed in the brain and skeletal tissues and appends a C18 acyl-CoA; CerS2 appends a C22-24 acyl-CoA and is ubiquitously expressed, though enriched in the liver and kidney; CerS3 appends C26-34 acyl-CoA and is expressed in skin and testes; CerS4 is expressed in skin, leukocytes, heart, and liver and adds a C18-20 acyl-coA; CerS5 and CerS6 both append a C14-16 acyl-CoA chain, but CerS5 is minimally expressed save for the skin, testes, and kidneys while CerS6 is expressed primarily in the intestine and kidneys95. The many chain length variants of Cer will be differentially modified into many variants of “higher” sphingolipids within the trans-Golgi apparatus, a process which occurs in tight coordination with Cer synthesis. Crucially, the total Cer content in the ER membrane is kept at trace levels by ORMDL proteins, which sense excessive Cer buildup and inhibit the SPT complex if excess Cer accumulates in the ER96. Many have previously shown that ER Cer accumulation triggers an apoptotic cascade, resulting in cell death97.
The Cer transport protein (CERT) mediates the transfer of Cer to the trans-Golgi. Here, a relatively small portion of Cer is modified with glucose, galactose, or lactose to produce a cohort of glyco-ceramides, further modified into products such as globosides, gangliosides, sulfatides, and many so-called glycosphingolipids (GSLs)94,98. However, the bulk of total Golgi Cer is converted into sphingomyelin (SM) by sphingomyelin synthase 1 (SMS1) by transferring a phosphocholine headgroup from phosphatidylcholine, producing diacylglycerol (DAG) as a side-product94. Intriguingly, this secondary role for SMS1 (producing DAG) is a primary mechanism of regulating DAG synthesis in the Golgi99. Among others, Cerbon et al. report that DAG produced by SMS1 regulates PKC activity and cell proliferation100.
Once synthesized, the vast majority of SM and glycosphingolipids are exported to the plasma membrane via secretory vesicular trafficking. These trafficking patterns are mediated by highly selective proteins that direct the newly synthesized lipids to the appropriate compartments in the cell. In 2007, Koivusalo et al. showed that the chain length of the secondary acyl chain of SM impacts its trafficking throughout the cell101. In support of this, in 2012, Contreras et al. identified an SM binding domain in the COPI sorting protein p24. This domain selectively binds one particular SM species, SM(d18:0/18:0), and mediates its transport to the plasma membrane102. Genetically transferring this binding domain to p23 confers SM binding capacity. Similarly, the protein FAPP2 transports glucosylceramide within the Golgi for further modifications103 and, as its name would suggest, the ceramide 1-phosphate transport protein mediates the transport of ceramide 1-phosphate between the Golgi and the plasma membrane104.
Most sphingolipids in the cell accumulate as SM or GSLs at the plasma membrane105. The glut of the remaining intracellular pool of sphingolipids is located in the endosomal compartment of the cell105. The endosomal SM primarily comprises lipids re-internalized from the plasma membrane during events such as phagocytosis101. The role of these endosomal SM and GSLs will be discussed extensively in Chapter 4. These intracellular sphingolipids are the primary source of the so-called “bioactive sphingolipids,” which are short-lived and induce potent cellular effects.
2.5.2 Sphingolipid breakdown: signaling through salvage
Freysz and Mandel showed in 1980 that the half-life of SM can be measured in weeks or years, depending on the tissue106,107 – in contrast, sphingosine 1-phosphate (S1P) has a half-life as low as fifteen minutes in the blood108. The shorter half-lives of sphingolipids such as S1P reflect the potency of their effect as signaling molecules. Perhaps the most well-characterized example of sphingolipid signaling, S1P is a potent chemotactic signal for lymphocytes: T cells express S1PR1 (one of five S1P receptors) and rapidly extravasate from the circulatory system to sites of tissue damage or infection up a gradient of S1P concentration109. Because lymphocytes must travel up an S1P gradient during chemotaxis, S1P in the plasma and healthy tissue must be kept at exceedingly low concentrations and produced in rapid excess in damaged or infected tissue. Given these potent effects and these short half-lives, one may ask: “Where do signaling sphingolipids come from?”
Of course, the cell can synthesize bioactive signaling lipids such as Cer and S1P via de novo synthesis, trafficking these products through the Golgi, and exporting them through secretory vesicular trafficking. Indeed, Menuz et al. and others have shown that de novo ceramide synthesis is upregulated in response to hypoxia and anoxia110,111. However, recent work has shown that the primary source of bioactive sphingolipids is the degradation of SM and GSLs at the plasmas membrane and within the endosomal compartment93,108. As depicted in the right side of Figure 2.2, SM can be sent back through the sphingolipid synthesis cascade to result in localized accumulation of Cer, Sph, and S1P94. Several sphingomyelinases (SMases) are known. Neutral SMase is most active at neutral pH and mainly resides in the outer leaflet of the plasma membrane (mainly cleaving extracellular SM). The highly related acid sphingomyelinase (aSMase) is regulated by acidic pH – this enzyme is primarily active within the lysosome. The degradation of lysosomal SM (and the subsequent production of sphingosine) induces targeted autophagy of the lysosome (lysophagy) and, subsequently, programmed cell death112. Additionally, cytosolic SMase activity has been shown to play a role in coordinating membrane repair mechanisms113,114. Similar glycosphingolipid-degrading enzymes can send glycosphingolipids back through the synthesis cascade to produce ceramide94.
Enzymes such as sphingomyelinase also allow cells to acquire extracellular sphingolipids and incorporate them as membrane constituents via salvage94. We have previously reported that ablating all forward sphingolipid synthesis (via genetic knockout or inhibition of the SPT complex) does not fully deplete a cell of all sphingolipids90 – and in fact, we anecdotally note that cells inhibited in sphingolipid production rapidly die when cultured in serum-free media.
Having described the sphingolipid metabolic network, we can discuss how sphingolipids are involved in disease before returning to their potential roles during Mycobacterium tuberculosis infection.
2.5.3 Sphingolipids and disease
Dysregulation of sphingolipid biology is intricately tied to many diseases. We and others have written extensively on this topic (Chapter 7)91,115,116, and this review will only briefly touch on some of the most well-characterized examples of diseases of sphingolipid dysregulation, as the scope of sphingolipid metabolism disorders encompasses every step of the sphingolipid pathway and could easily fill many volumes.
Several of the most studied sphingolipid disorders cause lysosomal storage disorder (LSD). A spectrum of more than 70 individual diseases, LSD is generally a progressive disorder that results in the accumulation of lysosomal contents117. The most common LSD is Gaucher disease, a genetic deficiency in the glucocerebrosidase (GBA) enzyme. GBA mediates the cleavage of glucosylceramide into glucose and ceramide, and the severity of the disease aligns with the activity of a patient’s GBA alleles117. Gaucher disease is characterized by developmental neurological delays, progressive loss of bone density, and cachexia117. Beyond Gaucher disease, GBA mutations are strongly associated with the neurodegenerative Parkinson’s disease118.
Similarly, Fabry disease is the product of mutations in the alpha-galactosidase (GLA) enzyme, which results in the accumulation of another glycosphingolipid, globotriasylceramide (GL3)117. Enzyme replacement therapy (ERT) is an effective treatment for many Gaucher and Fabry patients. In ERT, patients receive infusions of exogenous enzyme to complement the deficient endogenous activity117,119. During the production of these therapies, the recombinant enzyme is mannosylated – this encourages endocytic uptake and, conveniently, the trafficking of the enzyme directly to the site in which it is needed119. Ongoing efforts are underway to find delivery methods to treat the neurological manifestations of these diseases, as in ERT, the recombinant enzyme does not efficiently cross the blood-brain barrier118.
Beyond lysosomal storage disorders, sphingolipids are known to play intricate roles throughout development, aging, osteoporosis, and many cancers. The interface between host and pathogen is an intriguing and developing research arena. We have previously written on this topic (including several of the works presented in this dissertation: Chapter 827, Chapter 789), as have others. Utermolen et al. discussed in 2008 the role of aSMase as a target for several bugs, including Neisseria gonorrhoeae and Listeria monocytogenes43. These authors find that aSMase appears essential for enabling the many membrane fission/fusion events that must occur as the lysosome matures. We have published similar reports on the necessity of sphingolipid production for efficient phagocytosis of environmental pathogens (Chapter 3)90,120.
Notably, sphingolipids are largely absent from the prokaryotic lipidome - with the exception of a handful of bacterial phyla, including Bacteroides, Sphingomonas, and Sphingobacterium121–124. The many roles that these bacterial sphingolipids play in human health are discussed at length by Bai et al, 2023124 – these roles span from pro- to anti-inflammatory among commensal bacterial strains122,125. Here it must be noted that Mtb is not among the bacterial families which are capable of de novo sphingolipid synthesis – an in vitro, cultured Mtb bacillus is devoid of all sphingolipids; however, numerous reports have shown that Mtb does express a gene, Rv0888, which has demonstrable sphingomyelinase activity126, and which allows Mtb to utilize carbon from SM during replication127.
With this context, we can return to the roles of sphingolipids during Mtb infection.
2.6 Open questions regarding sphingolipids in Mtb infection
There are many open questions regarding how sphingolipids may influence the progression of a tuberculosis infection. We can categorize these questions into groups: (1) Does Mtb utilize host sphingolipids as a carbon source? (2) Does sphingolipid dysfunction in host cells impact Mtb survival/growth or TB disease progression? And, finally, (3) Can sphingolipid biology be modulated to serve as host-directed therapy? We will briefly summarize the current literature surrounding each of these questions below.
It is known that Mtb utilizes host lipids as a source of carbon and energy: this includes stimulating cells to produce triacylglycerides and sterols in high abundance and storing them in lipid droplets, and subsequently utilizing fatty acids released from these triacylglycerol and sterols by lipases; these fatty acids appear to be used in both fatty acid β-oxidation (energy production) and directly utilized in the synthesis of Mtb lipids69,87,128–131 Many fewer studies have investigated the utilization of host sphingolipids – however, as noted above, in 2015, Speer et al. identified the sole enzyme with SMase activity in the Mtb genome, Rv0888, and that the expression of this enzyme is dependent on the presence of SM and enables Mtb growth on SM-supplemented minimal growth media127. Further, Dang et al. showed that this enzyme is both membrane-bound and secreted and contributes to lung pathology by exacerbating neutrophil-induced inflammation132. Much work remains to fully understand whether other sphingolipids may similarly serve as energy sources for Mtb during infection. For instance, the above studies by Speer et al. and Dang et al. do not investigate GSLs or Cer as potential catabolites of Rv0888.
More work has sought to characterize the roles of sphingolipids in the host antimicrobial response to Mtb. In one example, Nadella et al. and others have reported that the potently bioactive sphingolipid sphingosine 1-phosphate (S-1P) is a crucial factor in stimulating the recruitment of T cells and macrophages to the site of Mtb infection and that high localized levels of this sphingolipid product stimulate potent M1 differentiation in macrophages – activating the production of inducible nitric oxide synthase (iNOS)133–135. Nadella et al. also showed that S-1P treatment enhanced control of Mtb infection in both in vivo and in vitro mouse models133. However, other studies have shown that direct S-1P treatment via aerosolization can induce hyperresponsiveness to bronchoconstriction – perhaps precluding its utility as an antitubercular therapeutic136. An alternative to direct S-1P would be to increase the activity of sphingosine kinase (SphK), which produces S-1P, or increase the abundance of sphingosine substrate for SphK, as discussed below.
In another example of sphingolipids modulating the severity/progression of infections, McGovern et al. showed that aSMase deficiency confers high susceptibility to infectious pneumonia137 and that active TB patients display lower aSMase activity135. Wu et al. found in 2020 that aSMase deficient mice are highly susceptible to infection by the attenuated Bacillus Calmette-Guerin (BCG) strain of M. bovis. Adoptive transfer of wildtype (aSMase+) macrophages into knockout mice (aSMase-) restores the ability to control BCG infection138. Vasquez et al. observe that Functional inhibitors of acid sphingomyelinase (FIASMAs), a common category antidepressant including amitriptyline and desipramine, have been shown to result in enhanced mycobacterial growth139. These reports on aSMase regulating the antimicrobial response to Mtb are intriguing because several mechanisms appear simultaneously enabled by aSMase activity.
Ceramide produced by aSMase has direct biological effects within the lysosome. In one example, Cer directly regulates proteases such as Cathepsin D (CTSD). While CTSD has been long known to be inhibited via membrane association140, Heinrich et al. showed in 2000 that CTSD activity is directly coupled to aSMase activity, and it has since been shown that direct binding of Cer stimulates autoproteolysis to release a 32 kD catalytically active CTSD protein from the lysosomal membrane fraction141,142. Others have similarly found that Cer binds to Cathepsin B143, phospholipase A2144, and the kinase suppressor of Ras-1145. Cer’s activation of these and many other proteins is at least partially related to this lipid’s self-association into Cer-enriched platforms146,147. As described above, Cer produced by aSMase induces direct cellular effects and also serves as the primary cellular source for other signaling sphingolipids, such as sphingosine 1-phosphate.
As a product of the above mechanisms, aSMase activity has been tightly linked to the regulation of cell death pathways – namely, the decision between inflammatory necrotic cell death and non-inflammatory apoptotic cell death. Lysosomal SM-derived Cer serves as a second messenger that induces translocation of BAX from the cytosol to the mitochondria, inducing loss of mitochondrial integrity and cytochrome c release148. In parallel, it has been long known that mycobacterial infection appears to manipulate this pathway to induce non-inflammatory necrotic cell death149. The subtlety between routes of cell death that promote Mtb control versus those that enable unrestricted Mtb growth is yet to be fully characterized – though this has been the subject of many studies: Butler et al. review contradicting evidence by several studies, and they report that mycobacterial virulence is not a factor on necrotic versus apoptotic cell death150–153. Bringing a long point home: there are many open questions regarding how aSMase activity may be a critical inflection point in the success or failure of an Mtb infection, and much work remains to explore the possibility of modulating this enzyme in a host-directed therapy.
As noted by Mohammed et al. in their 2022 review, the sphingolipid pathway is ripe with HDT possibilities135. However, the nuances of infection mean that different patients may require dramatically different paradigms of sphingolipid modulation, depending on their latent-vs-active TB state, lipid profile, and immunocompetence. Much work remains to uncover how sphingolipids influence the progression of an Mtb infection. Questions such as these have compelled the work in this dissertation. In Chapter 3, we will explore our findings that sphingolipids are essential for the initial stage of Mtb infection: entry into the host cell via phagocytosis. In Chapter 4, we will discuss the apparent role of sphingolipid synthesis in repairing endosomal/lysosomal damage induced by Mtb pathogenicity factors.
2.7 Challenges and breakthroughs in studying lipids
With all the evidence described above establishing sphingolipids as strong candidates for host-directed therapy, one must ask: why are there so many open questions about how SM, Cer, and S-1P affect Mtb infection? With the clear ties with cell survival and immune signaling, one would expect a concerted effort to address whether this pathway is amenable to HDT. However, studying lipids can be challenging, with logical pitfalls and technical challenges.
I describe many such challenges in studying sphingolipids during viral infections in detail in Chapter 789. In brief, however, lipids lie at the terminus of the central dogma – they are the product of a long series of chemical reactions mediated by proteins. One cannot use simple genetic tools (knockout or point mutations) to alter a lipid’s structure or trafficking subtly. Each lipid species is a part of a complex metabolic network with astounding potential to be modified and subsumed into other lipid classes or degraded into base components. The comparatively small size of most lipid species precludes changing their structure dramatically, and their hydrophobicity/biochemistry often precludes ectopic treatment. Similarly, their small size means there are few interfaces with which a lipid-binding protein can strongly interact; for example, few lipids can participate in hydrogen bonding. Finally, the biophysics and effects of specific lipids prevent simple complementation via add-back experiments.
Sphingomyelin is an excellent example of a challenging lipid to study directly. Add-back experiments with SM are nearly impossible because this conical lipid strongly prefers planar, rigid, impermeable membrane structures, and treating cells with SM-enriched micelles can only provide a low percentage of SM content. Some studies have even proposed SM/Cholesterol liposomes as slow-release encapsulations for drug/vaccine delivery154,155. The utility of encapsulating drugs in liposomes of this composition lies in the fact that SM-enriched membranes are notably recalcitrant to fuse with other membranes, allowing these encapsulations to stay in the plasma longer154,155. In contrast, ceramide can be fed to cells as an ectopic source of sphingolipids156. However, it is well-documented that excess Cer is a potent inducer of apoptosis157. The apoptosis-inducing effects of Cer have driven substantial research into using Cer-loaded liposomes as a chemotherapeutic against cancer156.
And, though this list is not comprehensive, a final challenge in studying lipids is that their metabolic network is a sprawling and complex architecture in which lipids are rapidly interconverted, modified, and catabolized. Blocking the synthesis of a particular lipid will result in accumulating its source inputs and losing any secondary products. For example, inhibiting sphingomyelin production results in ceramide accumulation, phosphatidylcholine accumulation, and a reduction in diacylglycerol production; the cell will partially respond to ceramide accumulation by reducing SPT activity, but it will also result in increased glycosphingolipid production158,159. In addition to these direct effects resulting from inhibiting the pathway, there can be secondary effects: blocking SMS activity also induces a significant loss of cellular cholesterol content158. Inhibiting or knocking out one enzyme is much like removing a node in a spider’s web – the whole network must adjust to the node’s absence.
These examples demonstrate that the reductionist approach has suited many aspects of biological study (observe the effects of gene knockout, then determine if reconstituting restores the status quo). With these challenges in mind, a question arises: “How does one study sphingolipids?” We have previously written extensively on genetic knockout to study the roles of sphingolipids (Chapter 7)89. Here, we will briefly touch on techniques that allow for measuring and visualizing native lipids and techniques that employ synthetic lipid analogs to track the metabolism and interactions of the target lipids.
As with the field of genomics and the advent of next-generation sequencing, the fields of proteomics and lipidomics have seen an explosive maturation in the sensitivity, throughput, and depth of coverage that a mass spectrometer can achieve. The major hurdle has rapidly become: “How do we glean information from these tens/hundreds of thousands of mass spectra?” Tools like LIQUID160 allow for rapidly identifying many lipid species from mass spectrometry data. Databases such as MycoMass also annotate many lipid species unique to mycobacteria. These improvements in computational tools allow for deeper exploration into the broad changes that occur during a complex event such as an Mtb infection.
However, even highly sensitive mass spectrometers have difficulty identifying and quantifying the presence of highly transient lipid species such as plasma membrane Cer or serum S1P. The speed and quality of sample preparation can have an immense impact on the concentrations measured for these lipids. Greene et al. recently reported a new technique for measuring plasma membrane Cer161.
Similarly, a panel of sphingolipid-binding proteins serves as valuable tools in visualizing the localization and density of a sphingolipid. For example, the fluorescent fusion proteins lysenin-GFP and Equinatoxin-GFP enable the visualization of sphingomyelin via fluorescence microscopy. Lysenin and Equinatoxin were produced from pore-forming toxins isolated from Eisenia fetida162 and Actinia equina163. Similarly, an anti-ceramide antibody allows one to visualize ceramide in fixed cells and tissue164,165.
In parallel to these techniques, which enable the detection and visualization of native sphingolipids, a variety of synthetic sphingolipid analogs allow for in-depth analysis of the trafficking, protein-lipid interactions, and metabolism of select lipid species. Chief among these are the trifunctionalized lipid analogs reported by Höglinger, Haberkant, Farley, and Schultz112,166–171. These lipid analogs will be discussed in great detail in Chapter 6.
2.8 Conclusion
After years of research, it is clear that lipids play a crucial role in the interaction between Mycobacterium tuberculosis and host cells. Recent studies have further highlighted the importance of sphingolipids in directing the course of Mtb infection, and there is much interest in exploring the potential of sphingolipids as targets for host-directed therapy. This area of research has implications for developing new antitubercular therapies and broader implications for understanding how sphingolipids enable mammalian immune activity. As we unravel the complexities of Mtb infection, it is clear that a multidisciplinary approach to understanding the role of lipids in infection will be essential for developing new treatments and improving human health.
2.9 Contributions
GG compiled the concepts discussed in this review and wrote the entirety of the text. FGT oversaw the preparation of this review.