6 An Affinity for Sphingolipids
Trifunctional lipid probes reveal protein-sphingolipid interactions during Mycobacterium tuberculosis infection
6.1 Abstract
Understanding the molecular interactions underlying Mtb’s capacity for immune subversion and hijacking of the host cell can provide a unique insight into potential new routes of antitubercular therapy. Because Mtb is a pathogen with a unique sensitivity to the host lipid compartment, and because sphingolipids are a lipid family with emerging roles in regulating antimicrobial functions in macrophages, we sought to characterize the sphingolipid interactome of an Mtb-infected cell. To this end, we utilized trifunctionalized lipid analogs as affinity handles for enrichment proteomics. We found that Mtb infection induces a significant reprogramming of proteins that interact with sphingosine. These changes included a downregulation of interactions among proteins involved in secretory trafficking and lysosomal maturation and an upregulation of interactions with cytoskeletal proteins. We further identify a candidate Mtb protein that selectively interacts with trifunctional sphingosine. This analysis is the first of its kind and represents a crucial step in characterizing how sphingolipids mediate the cellular response to intracellular infection.
To accompany the main body of this report, we present an interactive format to explore the results of this study at the following: Trifunctional Proteomics Shiny App.
6.2 Introduction
As discussed in Chapter 2, Mycobacterium tuberculosis (Mtb) is a globally significant human pathogen. In the face of spreading antibiotic-resistant strains of Mtb, there is a growing need for novel host-directed therapy (HDT). Given their extensive roles in regulating mammalian cell survival and apoptosis, we and others have proposed sphingolipids as a strong candidate for anti-tubercular HDT (Chapter 3 and Chapter 4)1,2. Following this line of study, we sought to address whether the interactions between sphingolipids and proteins in the host cell are potential intervention points in host-directed therapy.
Lipid dynamics are a significant determinant of Mtb biology, as discussed in Barisch and Soldati’s 2017 review3. We and others have shown that host lipids, such as diacyl- and triacylglycerides, and lipid storage structures, such as lipid droplets, are substantially upregulated in response to Mtb infection (Chapter 5)3. Previous authors have postulated that the sphingolipid pathway contains numerous potential targets for anti-Mtb HDT1,2,4. However, there are significant open questions regarding whether the interactions between sphingolipids and proteins in the host cell are potential intervention points in host-directed therapy. We use a trifunctionalized sphingosine analog to explore the interactions between sphingolipids and proteins during Mtb infection. We expect that the Mtb-host interface will be highly dependent on protein-lipid interactions – but, historically, few tools have existed which allow for identifying the protein interactors of a given lipid species.
The advent of trifunctionalized lipids has circumvented many of the long-standing challenges of lipid studies. These challenges have been discussed at length elsewhere5. However, they include 1. the small size and potent hydrophobicity of lipids, which preclude many chemical modifications and exogenous supplementation or add-back; 2. the rapid interconversion of lipid species along complex and convoluted metabolic pathways; and 3. the transience of interactions between lipids and proteins5–7. In particular, trifunctional sphingosine (tf-Sph) has enabled significant breakthroughs in studying the roles of bioactive sphingolipids in the cell. The native form of sphingosine is a transient and potent signaling lipid known to induce profound effects in the cell[8; ]. In 2015 and 2017, Höglinger showed that trifunctional sphingosine induces rapid calcium release from the lysosome and eventual lysophagy (targeted autophagy of the lysosome)7,9. Our recent report described a novel sphingolipid probe’s synthesis and biological behavior: trifunctional sphinganine (tf-Spa)6. Tf-Spa Trifunctional lipids allow for nuanced investigation into the protein interaction partners of select sphingolipid species in various biological contexts.
In the present study, we utilize trifunctional sphingosine to explore selective interactions between proteins and sphingolipids in human THP-1 macrophages during infection with pathogenic and attenuated strains of Mycobacterium tuberculosis. To assess the specificity of identified interactions, we compare the interaction partners of tf-Sph with those of several other previously reported trifunctional probes. We find that, at baseline, sphingosine interacts with several lipid-modifying proteins such as phosphatidylinositol transfer protein beta (PITPNB) and lysosomal proteins such as Cathepsin D (CTSD) and lysosomal Pro-X carboxypeptidase (PRCP), among others. When applied to the Mtb-infected cell, we find that tf-Sph experiences a dramatically altered interactome – the many up- and down-regulated interactions reflect altered lysosomal activity, protein synthesis, and histone modifications, among others. We additionally identify modest enrichment of a single Mtb protein to tf-Sph, the uncharacterized Rv3466. We hope that revealing these alterations to the sphingosine-protein interactome will reveal novel avenues for manipulating the host-pathogen interface during Mtb infection.
6.3 Results
6.3.1 Trifunctional lipid probes offer unique utility in assessing functions, localization, and interaction partners of a given lipid species
The lipid biologist’s toolbox was recently greatly expanded by adding trifunctional lipid analogs (trifunctional probes). These synthetic molecules maintain high structural similarities to their natural counterparts but have been modified with three chemical domains that allow for nuanced and tightly controlled dissection of a given lipid’s cellular trafficking, metabolism, and interactions (Figure 6.1). We and others have previously reported on their synthesis and utility, including Farley et al.6, Haberkant et al.10–12, and Höglinger et al.7,9. While many such trifunctional probes are now available, we consigned our focus to four probes in this study: trifunctional-Sphingosine (tf-Sph), tf-Sphinganine (tf-Spa), trifunctional-8:3-Fatty acid (tf-8:3-FA), and trifunctional-1:10-Fatty acid (tf-1:10-FA), as previously described by Farley et al. in 20236.
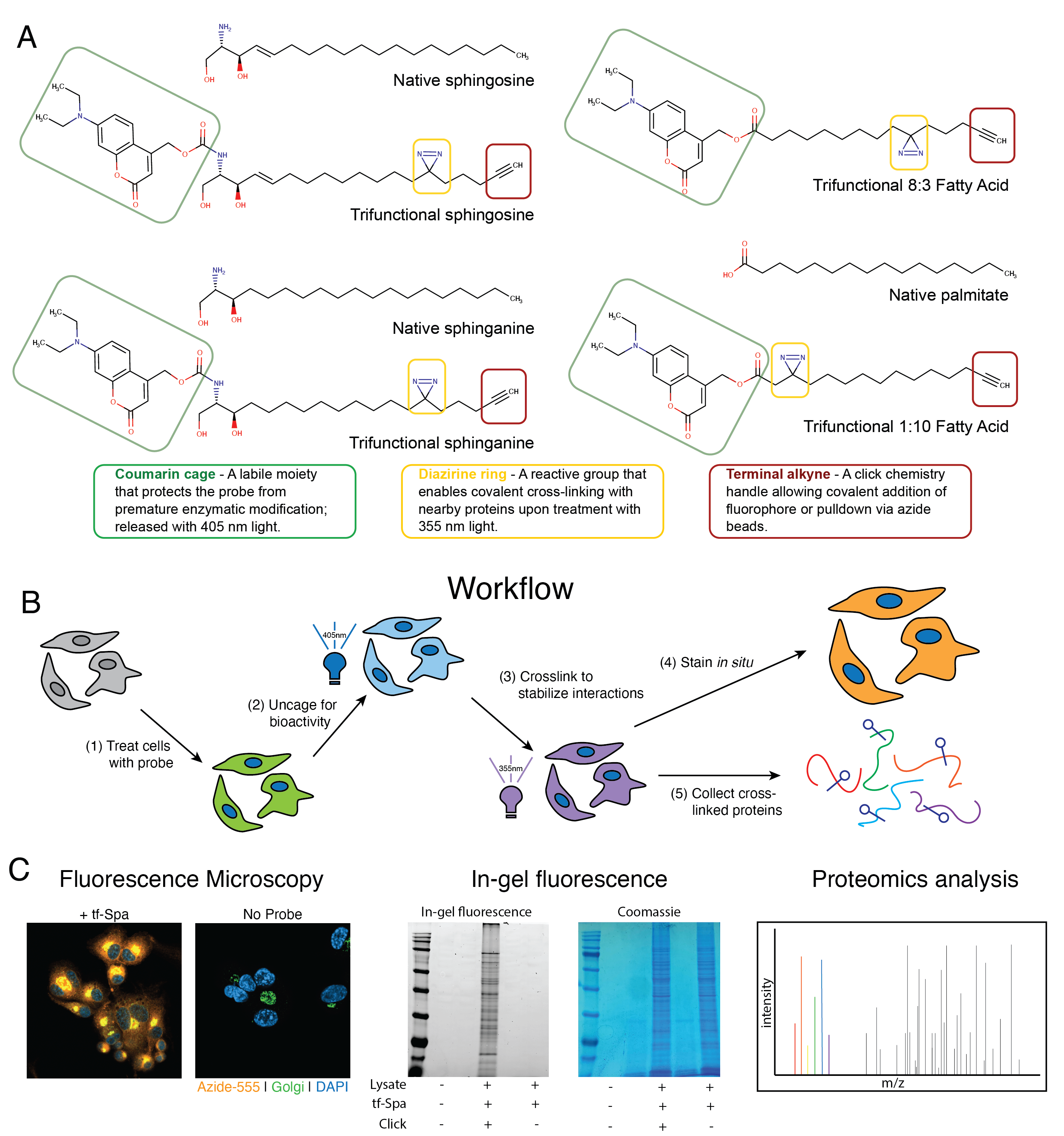
Each of these probes is expected to elicit a distinct cellular response, traffic to a distinct cellular compartment, and interact with a distinct cohort of proteins. For example, the two fatty acid probes differ only in the position of the diazirine ring: tf-8:3-FA is expected to preferentially bind to proteins that penetrate deep into the membrane, such as transmembrane domain-containing proteins. In contrast, the tf-1:10-FA probe would be expected to enrich more transiently membrane-associated proteins because the diazirine crosslinking motif is nearer to the hydrophilic headgroup. We expect these fatty acid probes to be incorporated into a wide array of lipids, including phospholipids, glycerolipids, and sphingolipids via transacylation.
In contrast, the trifunctionalized sphingolipid probes are expected to be incorporated into the sphingolipid metabolic pathway primarily. These probes differ only in a single double carbon bond at the C4-C5 position: sphinganine (or dihydrosphingosine) is produced early in the sphingolipid synthesis pathway and is incorporated into dihydroceramide by the ceramide synthases before being desaturated by dihydroceramide Δ4-desaturase13 and rarely accumulates in a typical cell. In contrast, sphingosine is a well-characterized signaling lipid produced primarily via the degradation of ceramide in response to signaling stimuli13. The production of sphingosine stimulates profound immediate effects in the cell9 and can also be used in ceramide synthesis as a part of the sphingolipid salvage pathway13. We previously reported on the subcellular localization of both trifunctional sphinganine and sphingosine6. In this report, Farley found that both probes are trafficked through the endoplasmic reticulum and the Golgi, aligning with the well-described sphingolipid synthesis pathway6,13. In the same report, Farley et al. also showed that tf-Sph retains its biological fidelity: it induces lysosomal calcium release, as previously shown9. Notably, however, tf-Spa fails to induce calcium release. These results indicate that tf-Sph retains its activity as a signaling lipid, whereas tf-Spa is primarily a sphingolipid precursor. We thus expect that the products of tf-Spa metabolism will interact with many proteins and that tf-Sph will initially interact with proteins involved in lipid signaling before being trafficked to the ER6,10.
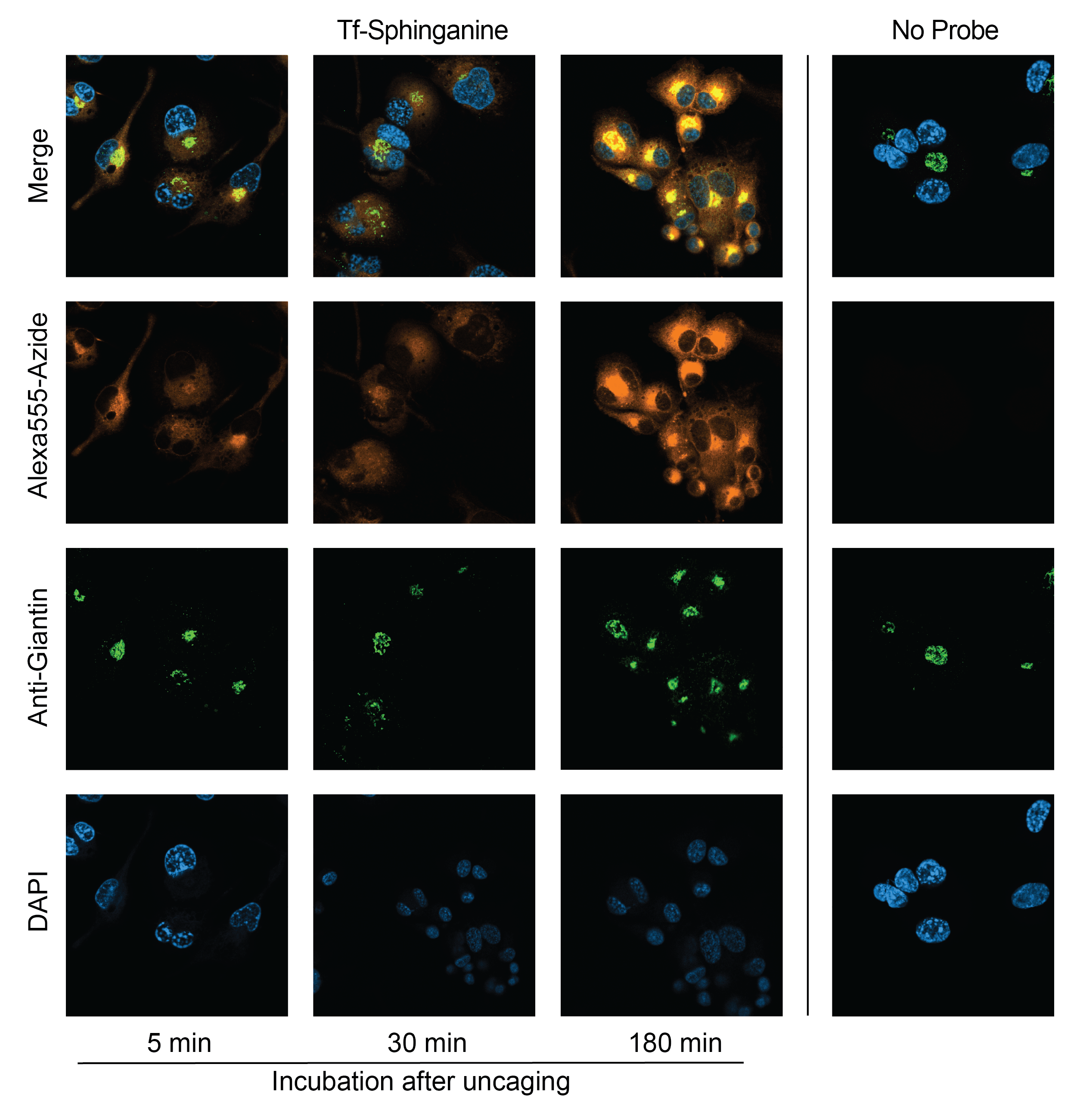
To validate these trafficking patterns in THP-1 cells, we visualized the subcellular localization of tf-Spa in THP-1 cells at increasing times after photo-uncaging (Figure 6.2). We observe similar patterns in these cells as has been previously reported by Farley et al.: tf-Spa experiences a time-dependent enrichment to the Golgi apparatus. These images serve primarily as validation of the utility of trifunctional probes.
6.3.2 Trifunctional probes reveal protein-lipid interactions of the THP-1 macrophage
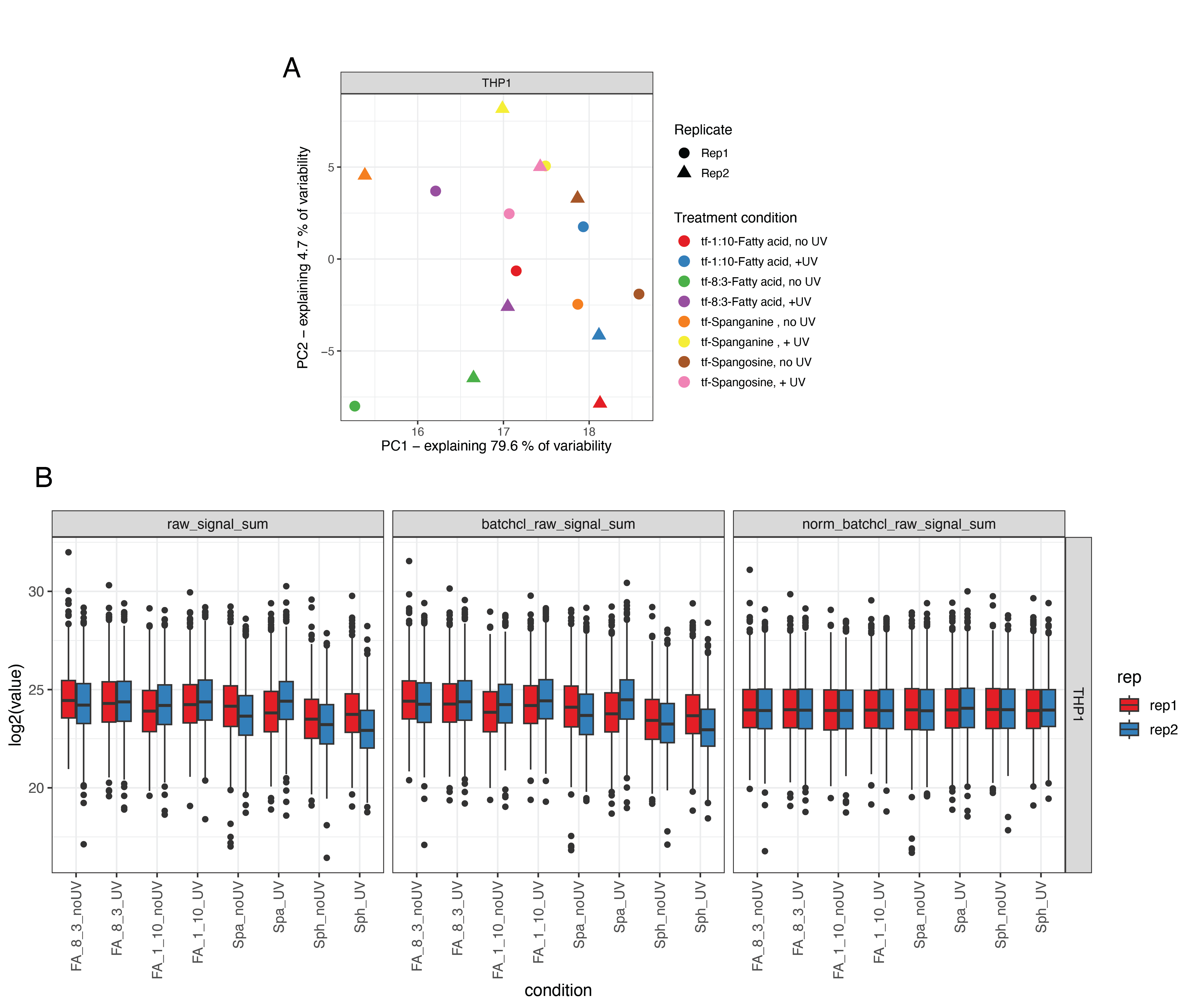
To identify the proteins that selectively interact with sphingolipid precursors in activated THP-1 macrophages, we performed an initial proteomics analysis to determine the protein interaction partners of the four trifunctionalized lipid probes: tf-Sph, tf-Spa, tf-8:3-FA, and tf-1:10-FA. Previous studies have used these or similar probes in human cervical adenocarcinoma cells (HeLa) and human liver epithelial cells (Huh7)6,7,9. However, human monocyte-derived macrophages (THP-1) have not been subjected to such lipid interaction analysis. Given that macrophages have dramatically distinct roles from epithelial cells and adenocarcinoma cells, we expected to identify a unique cohort of lipid-interacting proteins in these cells – including, presumably, (immuno)proteasome subunits, lysosomal resident proteins, and proteins involved in free radical production. The probe treatment and affinity enrichment procedures are depicted in Figure 6.3A and described in detail in Methods Section 6.7.3. All treatment and sample preparation were performed as previously described6,7,10. As a negative control, we prepared samples treated with each probe and uncaged but not treated with UV (-UV) and thus lacked crosslinking-stabilized interactions during enrichment.
We performed quantitative proteomics analysis using the TMT-16 isotopic labeling platform and LC-MS/MS. PCA analysis showed a distinct separation between +UV and -UV samples for each probe (Figure S6.1 A). We used variance stabilization to normalize the raw signal intensity of each channel (Figure S6.1 B). We identified 869 unique proteins with at least two unique peptides in at least two replicates. To identify the fold-change in the enrichment of proteins via diazirine crosslinking, we took the +UV versus -UV ratio for each probe and log2 transformed (Log2 fold-change, Log2FC). These results are depicted in Figure 6.3B.
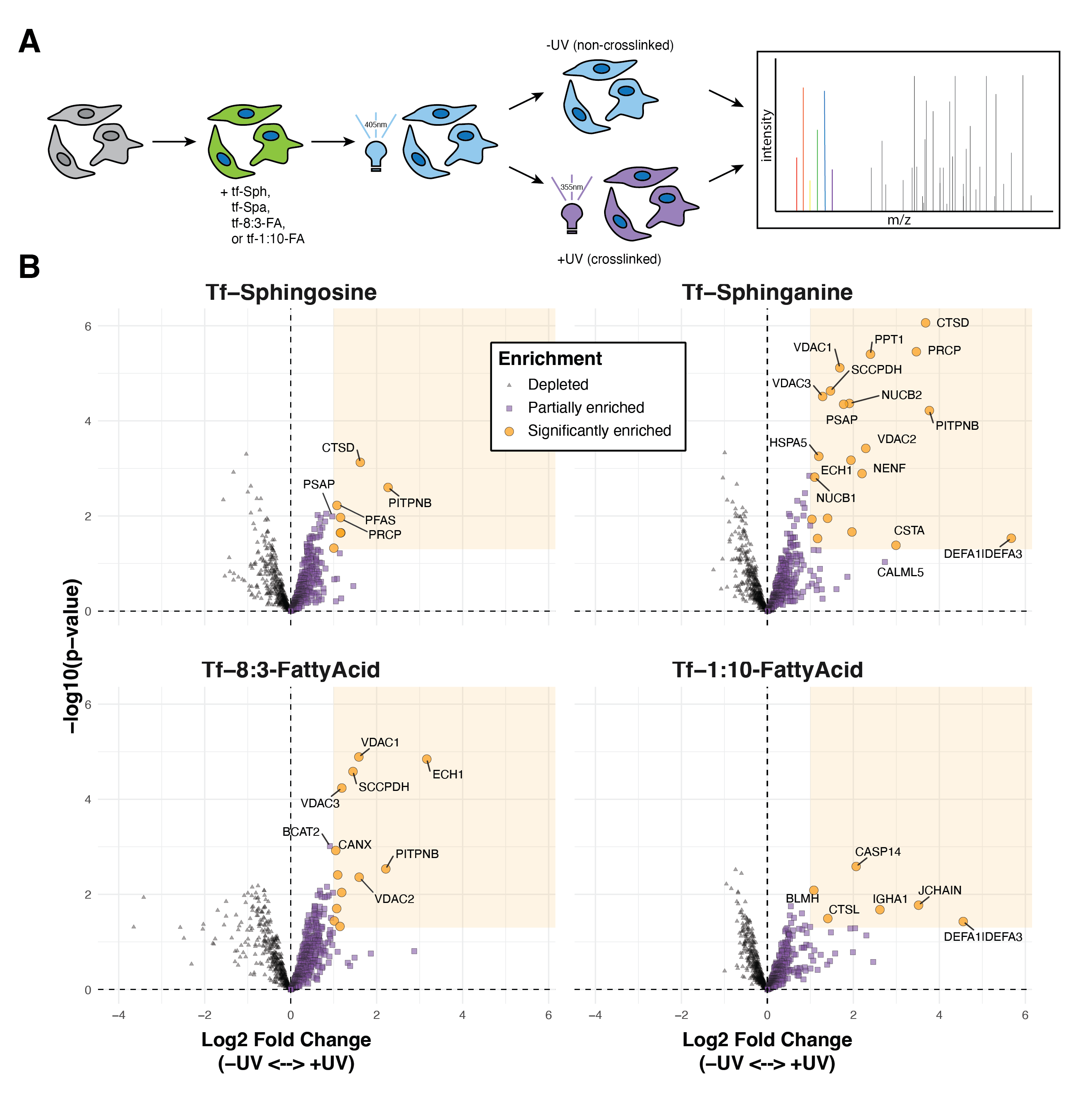
We applied Limma analysis14 to attain statistical significance for each protein between the +UV and -UV conditions. We applied an arbitrary cutoff for significance: LogFC > 1 and p-value < 0.05. Using these significance thresholds, we found that UV-crosslinking induced the enrichment of six proteins to the tf-Sph probe, twenty proteins to the tf-Spa probe, eight proteins to the tf-8:3-FA probe, and six proteins to the tf-1:10-FA probe. The relative enrichment of each significant protein among each probe (+UV versus -UV) is depicted in Figure 6.4A. Some proteins, such as VDAC2 and PITPNB, are enriched to more than one probe; other proteins, such as ATP6V1A, are uniquely enriched to a single probe.
We manually clustered each of the 35 enriched proteins into the following groups: proteins containing transmembrane domains, proteins with reported membrane association, secreted proteins, and proteins localized to the mitochondria, nucleus, or ribosome. Many of these putative lipid-interacting proteins are transmembrane domain-containing or peripheral membrane proteins. Relatively few are cytosolic or secreted.
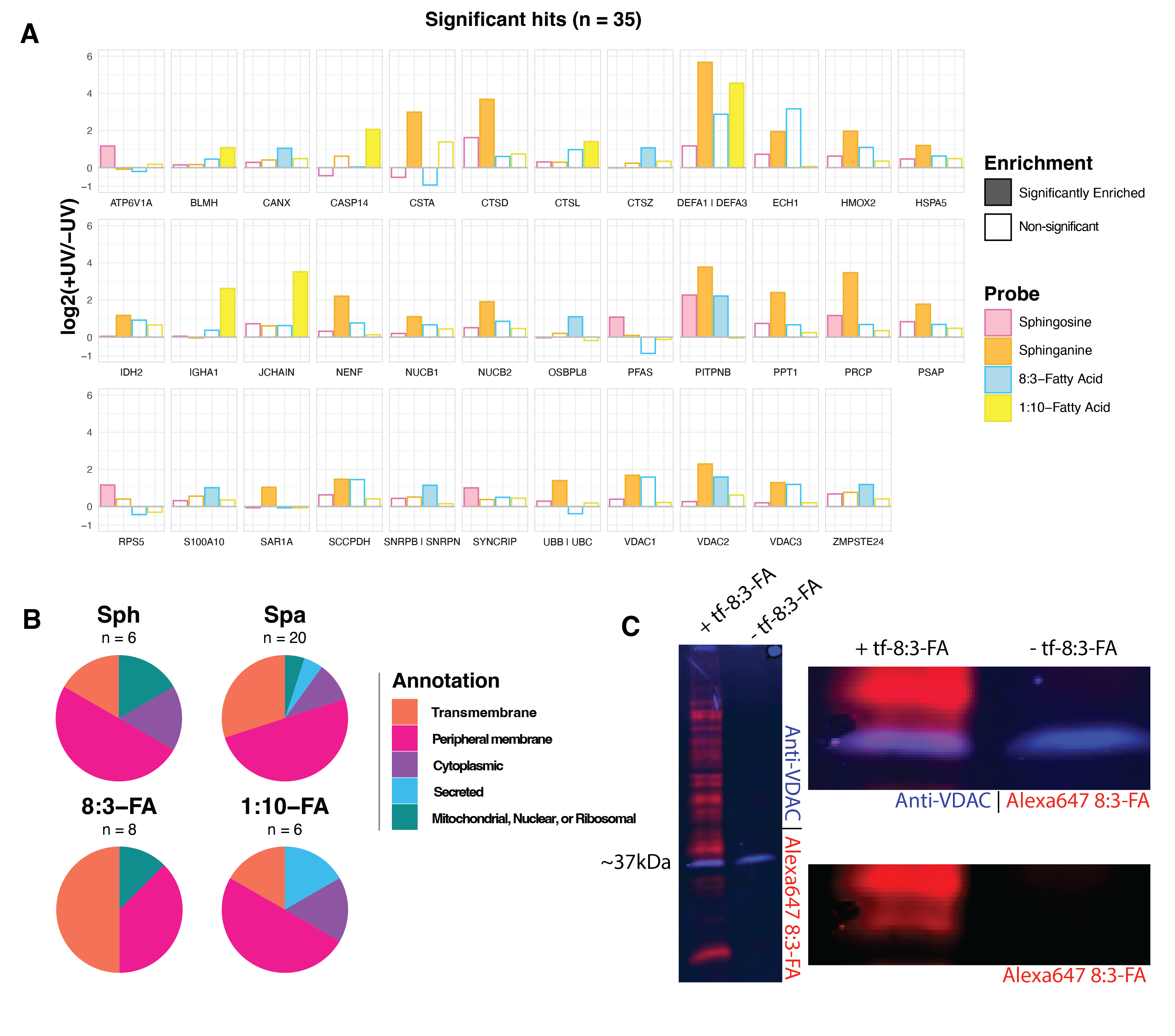
Our recent report investigated the sphingosine and sphinganine interacting proteins in Huh7 cells and characterized the similarities and differences between these two probes6. For both probes, we find significant overlap among significantly enriched hits. In the case of tf-Sphinganine, proteins such as NUC1 and NUCB2 (also named HEL-S-109), Cathepsin D (CTSD), ECH1, PITPNB, VDAC 1 and VDAC2 were enriched in both Huh7 and THP-1 cells. In contrast, we see several proteins that appear to be unique to THP-1 monocytes compared to previously reported interactors; these include Prosaposin (PSAP) and Neudesin (NENF).
Among these enriched hits are several interesting proteins with previously reported lipid interactions. Among these are the mitochondrial voltage-dependent anion channel isoforms VDAC1, VDAC2, and VDAC3. Previous studies have shown that VDAC2 contains a bona fide ceramide binding site and that interaction with ceramide is a potent trigger of apoptosis15. Further, Jahn et al. showed that VDAC1 and VDAC2 form a heterodimer which acts as a scramblase that transfers phospholipids synthesized in the ER into the inner mitochondrial membrane16.
Notably, these enriched proteins include several Cathepsin aspartic peptidases. Cathepsins A and D are enriched to both tf-Sphingosine and tf-Sphinganine. Cathepsins L and Z are enriched to tf-8:3-FA and tf-1:10-FA, respectively. There are many reported interactions between Cathepsins and sphingolipids. For example, CTSB is activated by lysosomal ceramide generated via aSMase, a process involved in NK and T cell apoptosis17. Similarly, sphingolipids are known regulators of CTSD activity18,19.
We also note the enrichment of Prosaposin to sphinganine and sphingosine. PSAP is a precursor polypeptide and is proteolytically cleaved into four small heat-stable glycoproteins: Saposins A, B, C, and D20. Each of these subunits is approximately 80 amino acids, with conserved glycosylation sites and identically placed cysteines20,21. Each Saposin appears to bind to a distinct cohort of higher sphingolipids. For example, Saposin A binds to glycosphingolipids, including glucocerebroside and galactocerebroside – and further, Saposin A stimulates acid β-glucosidase20. aSMase activity is regulated by the binding of Saposin22. Notably, Saposins have been shown to play critical roles in regulating lysosomal activity and immune function. Confusingly, there appears to be a cyclic regulation between Cathepsins and PSAP23. Cathepsins are the primary lysosomal peptidases responsible for cleaving PSAP into the mature Saposin subunits, and yet Saposins are widely reported for their capacity to stimulate CTSD activity18,23. What comes first? We find it intriguing that, in this proteomics analysis of the baseline interactions in THP-1 cells, we see “significant” enrichment of PSAP in sphinganine – however, PSAP was just below the Log2FC significance threshold.
These results reveal a diversity of lipid-interacting proteins in THP-1 monocyte-derived macrophages. Ongoing work seeks to validate and clarify these interactions’ biological impacts within the cell. For the remainder of this study, we used these results to inform a subsequent proteomics analysis on THP-1 macrophages infected with pathogenic and attenuated strains of Mycobacterium tuberculosis.
6.3.3 Proteomics of trifunctional probes reveals protein-sphingolipid interactions during pathogenic Mtb infection
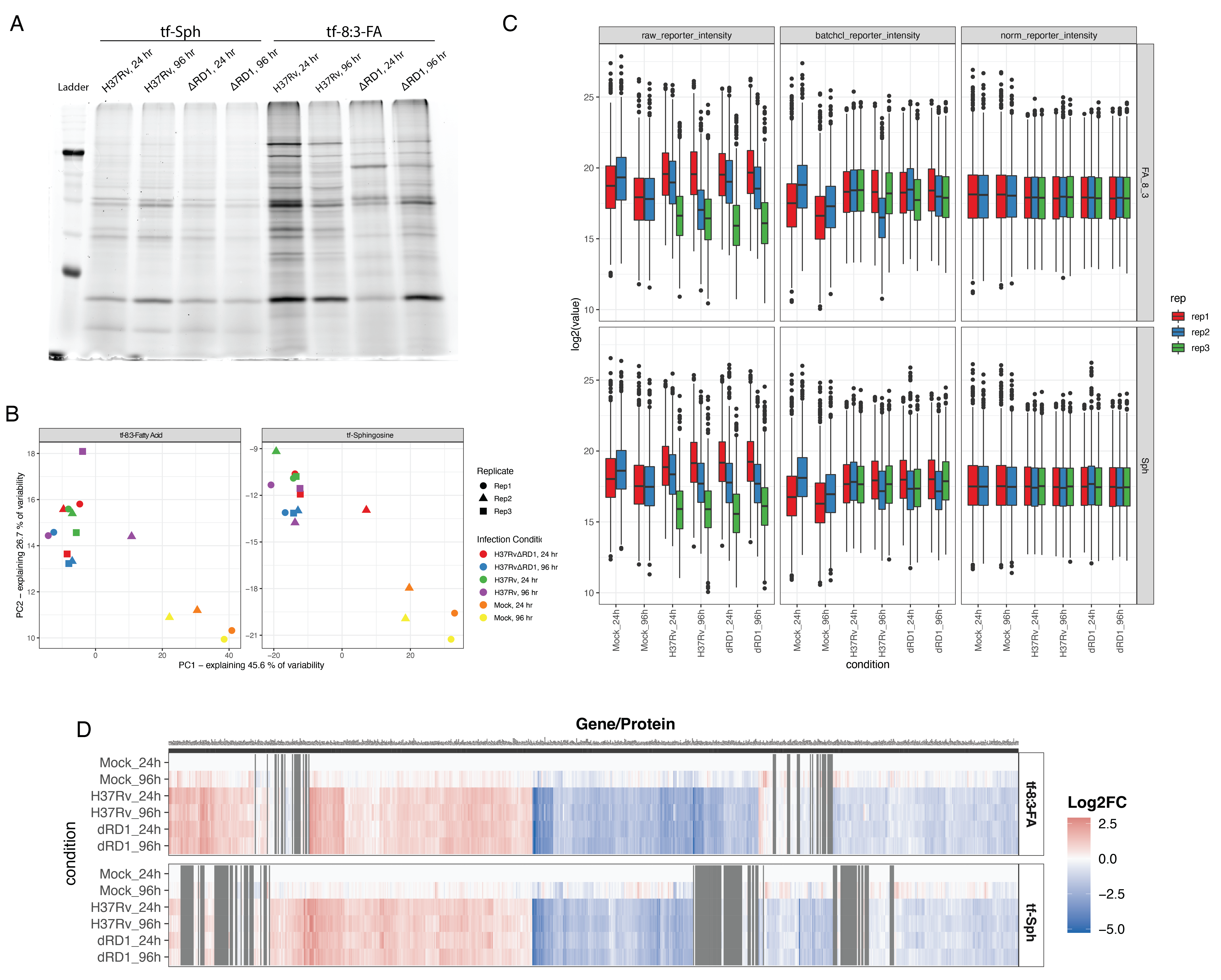
The main goal of this study was to analyze sphingosine interactions during Mtb infection. To achieve this, we infected cells with the pathogenic Mtb strain H37Rv for 24 and 96 hours, as depicted in Figure 6.5. Different multiplicities of infection were used for the two time points to maximize the infection rate at 24 hours and minimize cell cytotoxicity at 96 hoursFigure S6.3. At the desired time points, cells were treated with tf-Sphingosine as described in Section 6.7.3. We prepared a parallel experiment using the tf-8:3-Fatty acid probe as a control for non-specific lipid-binding proteins. After UV-induced crosslinking, cell lysates were collected, and a portion was used for in-gel fluorescence to validate sample quality (Figure S6.4 A). The remainder of each sample was clicked to azide-coated beads, and lipid-conjugated proteins were enriched. Samples were digested off the beads using trypsin and labeled with TMT-16 mass tags for LC-MS/MS proteomics analysis (one full TMT-16-plex for each probe). As above, data were normalized using variance stabilization (Figure S6.4 C). PCA analysis shows a wide separation between infected and uninfected samples for both lipid probes (Figure S6.3 B).
In total, there were 1169 unique proteins identified in the tf-Sphingosine-treated samples and 1411 unique proteins identified in the tf-8:3-FA-treated samples. Of these, 1039 were identified in both experiments, and 502 were identified in only one. We calculated the log2 fold-change of each protein for each possible comparison within TMT-16-plexes(Figure S6.5). We again performed statistical testing using Limma analysis. However, for this experiment, we applied a more stringent threshold for significance: a log2 fold-change > 2, a false-discovery rate < 0.05, and a p-value < 0.01. The enrichment annotation of the baseline UV experiment was applied to the proteins identified in this infection experiment. As a notable exception, we included PSAP as a “Significant” protein in our UV annotation, as discussed below.
| Probe | Infection (versus mock) | Timepoint | # Hits enriched | # Hits depleted |
|---|---|---|---|---|
| tf-Sph | H37Rv | 24 hrs | 31 | 120 |
| tf-Sph | H37Rv | 96 hrs | 35 | 109 |
| tf-Sph | H37Rv-ΔRD1 | 24 hrs | 19 | 78 |
| tf-Sph | H37Rv-ΔRD1 | 96 hrs | 35 | 101 |
| tf-FA | H37Rv | 24 hrs | 30 | 140 |
| tf-FA | H37Rv | 96 hrs | 23 | 86 |
| tf-FA | H37Rv-ΔRD1 | 24 hrs | 34 | 149 |
| tf-FA | H37Rv-ΔRD1 | 96 hrs | 25 | 118 |
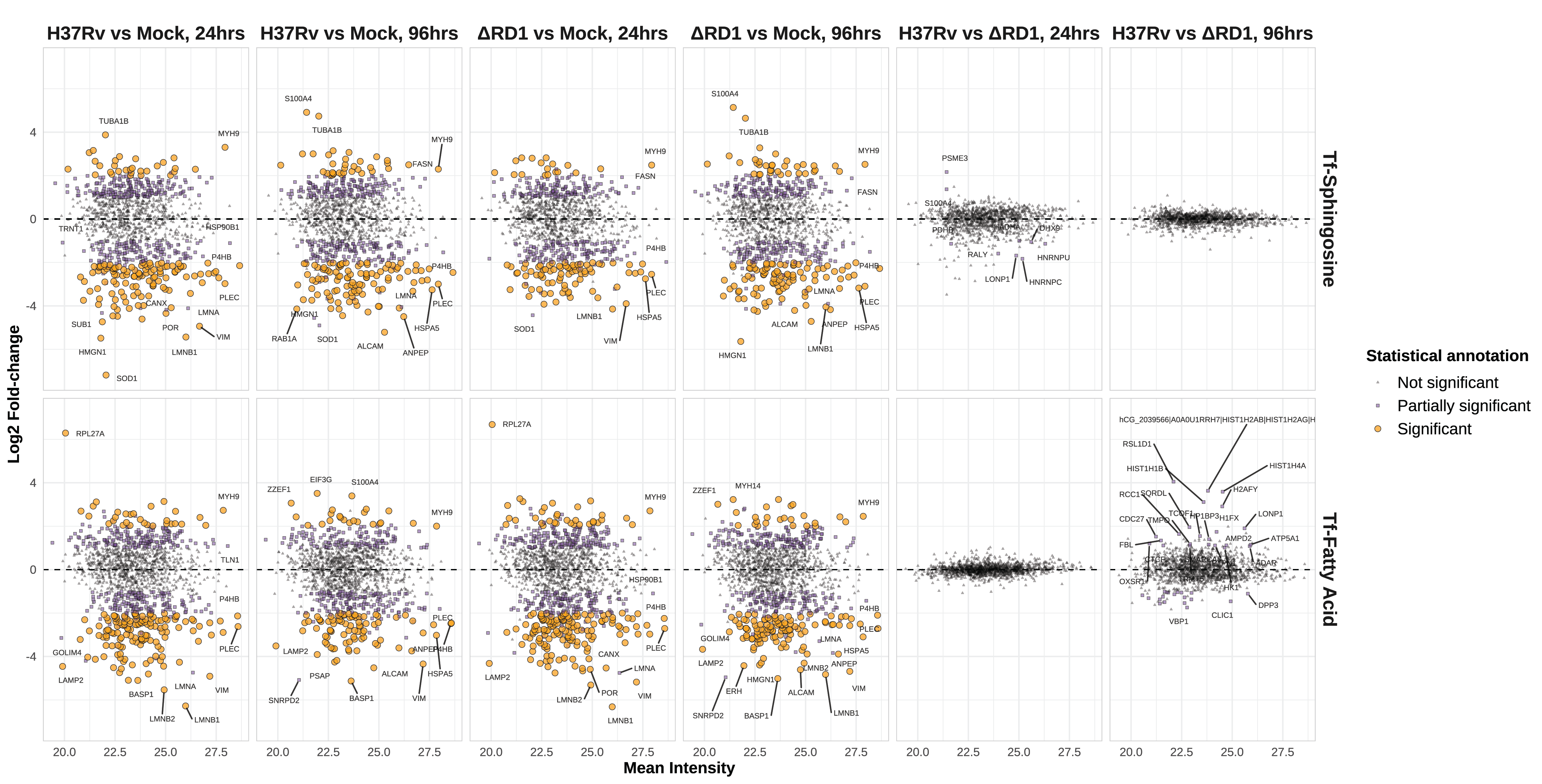
In assessing these data, there are several initial observations to be made. First, we see many statistically “significant” differentially enriched proteins for infection versus mock comparisons (Table 6.1). There is a propensity for decreased interaction with trifunctional probes, as many more proteins are significantly depleted during infection.
We must emphasize that these results represent the relative abundance of each protein after affinity enrichment. While this measure is intended to reflect the rate of interaction between each protein and the trifunctional probe, it also encompasses changes in abundance that result from altered transcription, translation, trafficking, and degradation of each protein. To determine whether the transcriptional response to infection impacted these results, we cross-referenced our results with the 2023 findings of Jani et al., who investigated the transcriptomes of cells infected with H37Rv (Figure S6.6)24. Several proteins are concomitantly enriched between Jani et al.’s data and our own (e.g., the interferon-induced gene MX1). Notably, however, most significantly downregulated interactions among our findings were not significantly changed at the transcription level. Public data on the whole cell proteome of H37Rv infected cells are not publicly available, and we cannot comment on the overall protein abundance for our differentially enriched hits.
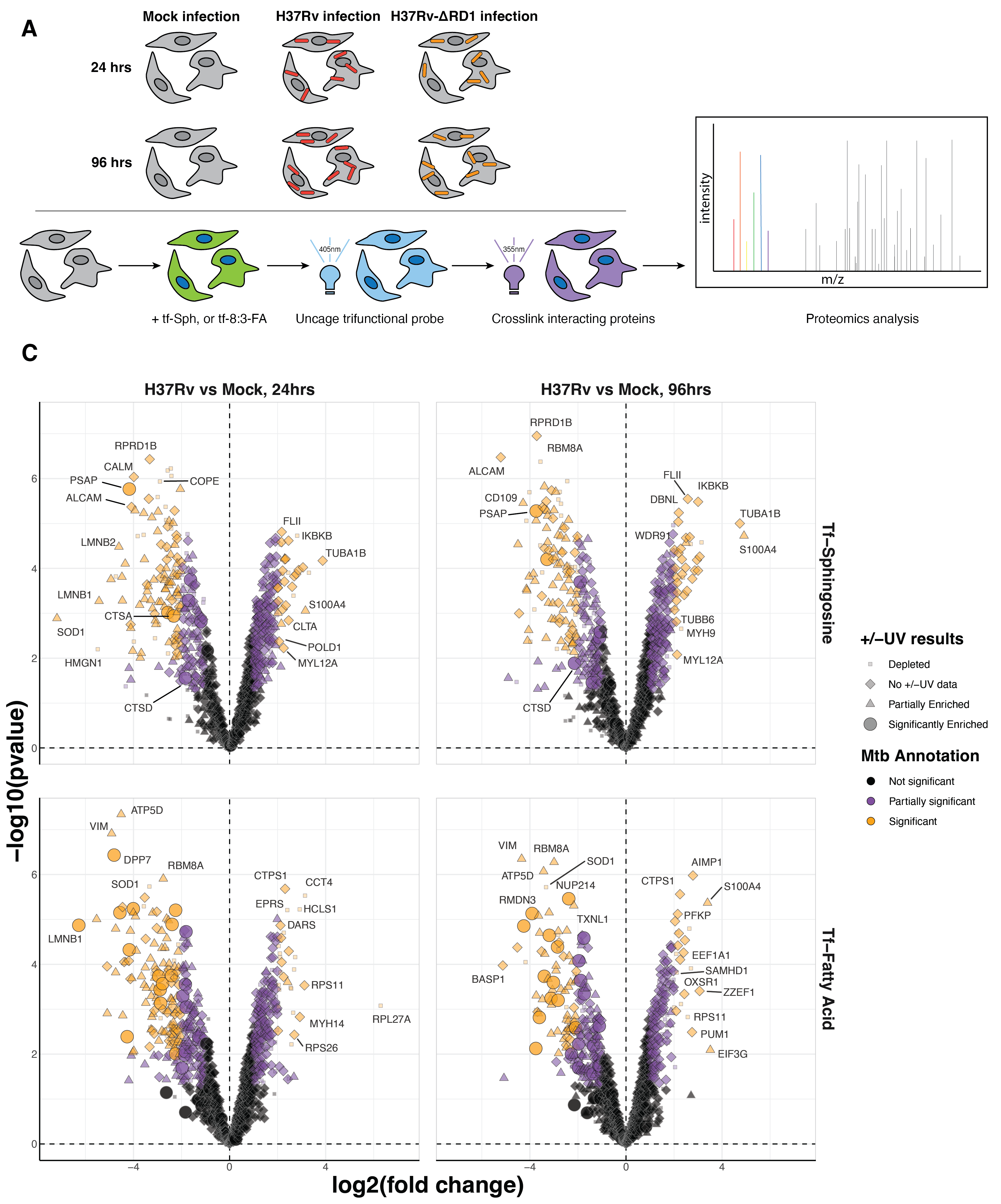
Focusing on 24-hour H37Rv infection, we observe several immunity-related genes among the protein interactions enriched during infection. These include MX1, IKBKB, and CTLA. We also observe several proteins related to motility and cytoskeletal trafficking, including Kinesin light chain 1 (KLC1), Tubulin, FLII, Microtubule-associated protein 4, Coronin-7, Myosin heavy chains 9 and 14 (MYH9 and MYH14), and Myosin light chain 12A (MYL12A). Intriguingly, we see a depletion of the activated leukocyte cell adhesion molecule (ALCAM), which is responsible for the extravasation of monocytes across the blood-brain barrier25. Some evidence suggests that BCG infection induces a transcriptional downregulation of ALCAM in PBMCs26. However, we do not observe this in our comparison to recent transcriptomics data reported by Jani et al.24.
Among the downregulated interactions, we see a preponderance of proteins in the secretory/endosomal compartment. These include the Coatomer subunit epsilon (COPE), GOLGA2, Vescicle-associated membrane protein-associated protein A (VAPA), Rab11b, PSAP, Cathepsin A, Cathepsin B, and Cathepsin Z (and Cathepsin D, though it was only partially significant). We see many cytoskeletal proteins among the upregulated interactions, including Tubulin, FLII, Myosin-9 and Myosin-14, Microtubule-associated protein 4, and Coronin-7. We postulate that these results indicate dysregulation of endosomal trafficking.
At 24 hours post-infection, we observe differential enrichment of several proteins of high interest in the secretory/endosomal compartment. Among these are Cathepsins A, B, and Z (CTSA, CTSB, and CTSZ; and CTSD, though it was only partially significant), Prosaposin (PSAP), Coatomer subunit epsilon (COPE), Vescicle-associated membrane protein-associated protein A (VAPA), Rab11b. We also see several calcium-binding proteins among these downregulated interactions: Calumenin, Reticulocalbin-1, and Calmodulin-1 (CALM1).
As noted above, there are prior reports of Prosaposin being involved in Mtb restriction, including the 2020 report by Shepelkova that showed that Saposin D is essential in the restriction of Mtb growth27. An intriguing hit is PH4P, which is significantly depleted at 24 hours. There are many reported functions of the protein P4HB in the cell. Among these, it is a structural subunit of the microsomal triacylglycerol transfer protein (MTTP). MTTP complex loads triglycerides, cholesteryl esters, and phospholipids onto apolipoprotein B (apoB)28. This complex is essential for presenting lipid antigens to NK T cells via CD1d29. In 2014, De Libero and Mori discussed that lipid antigens are essential for the invariant Natural Killer T cell response during Mtb infection30.
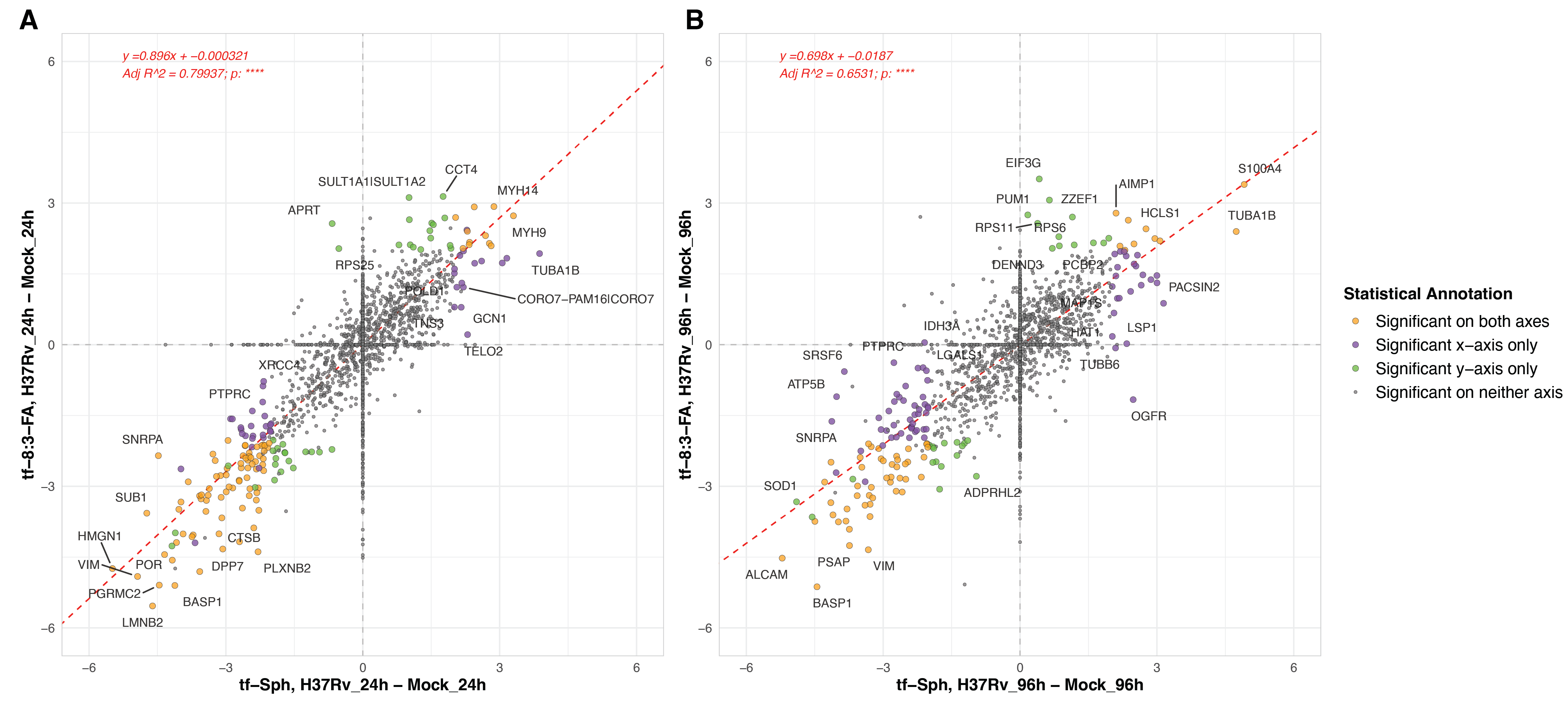
In comparing the enrichment of proteins to the tf-Sph and the tf-8:3-FA probe, we find a high degree of alignment among the co-identified protein at both time points (Figure S6.7). Linear regression analysis shows an adjusted R2 value of 0.80 for the 24-hour comparisons versus mock infection, suggesting a robust correlation between the datasets. More analytical work will be needed to distinguish proteins that interact more robustly with tf-Sph than tf-8:3-FA.
6.4 Pathogenic versus attenuated Mtb infections induce highly similar changes to the tf-Sph interactome
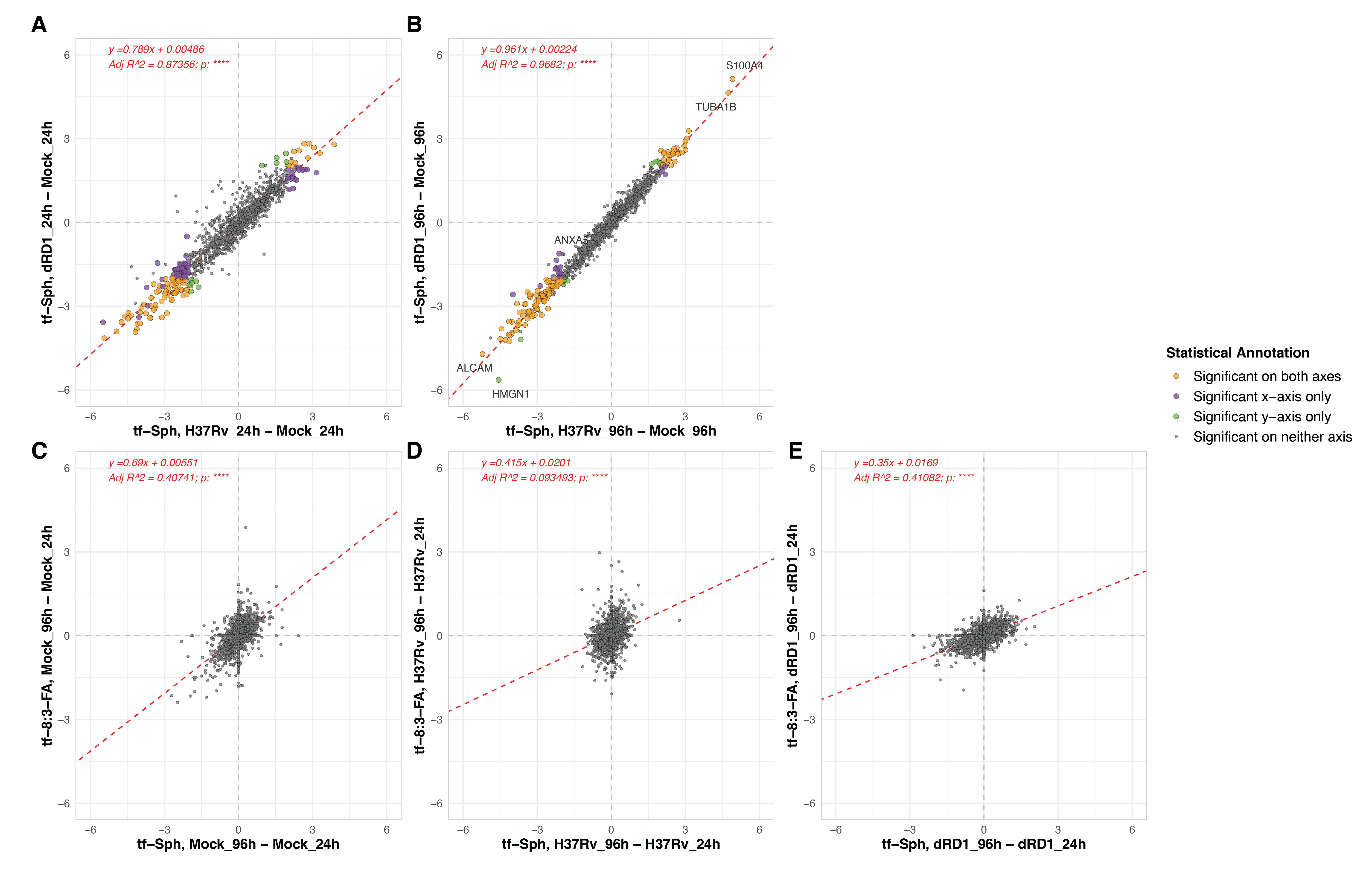
A primary goal of this study was to compare the effect of pathogenic and attenuated Mtb infections on the sphingosine interactome. To this end, we prepared parallel samples of cells infected with the attenuated Mtb strain H37Rv-ΔRD1 (this strain was discussed extensively in Chapter 5). In comparing the protein interactors of tf-Sph and tf-8:3-FA in these ΔRD1-infected cells versus mock-infected cells, we see many of the same hits as were differentially enriched in the H37Rv versus mock comparison (Figure S6.8). This is particularly evident when the log2 fold-changes of H37Rv versus mock are plotted against those of H37Rv-ΔRD1 versus mock – linear regression shows that these datasets have an adjusted R2 value of 0.87 (Figure S6.9 Upper panels).
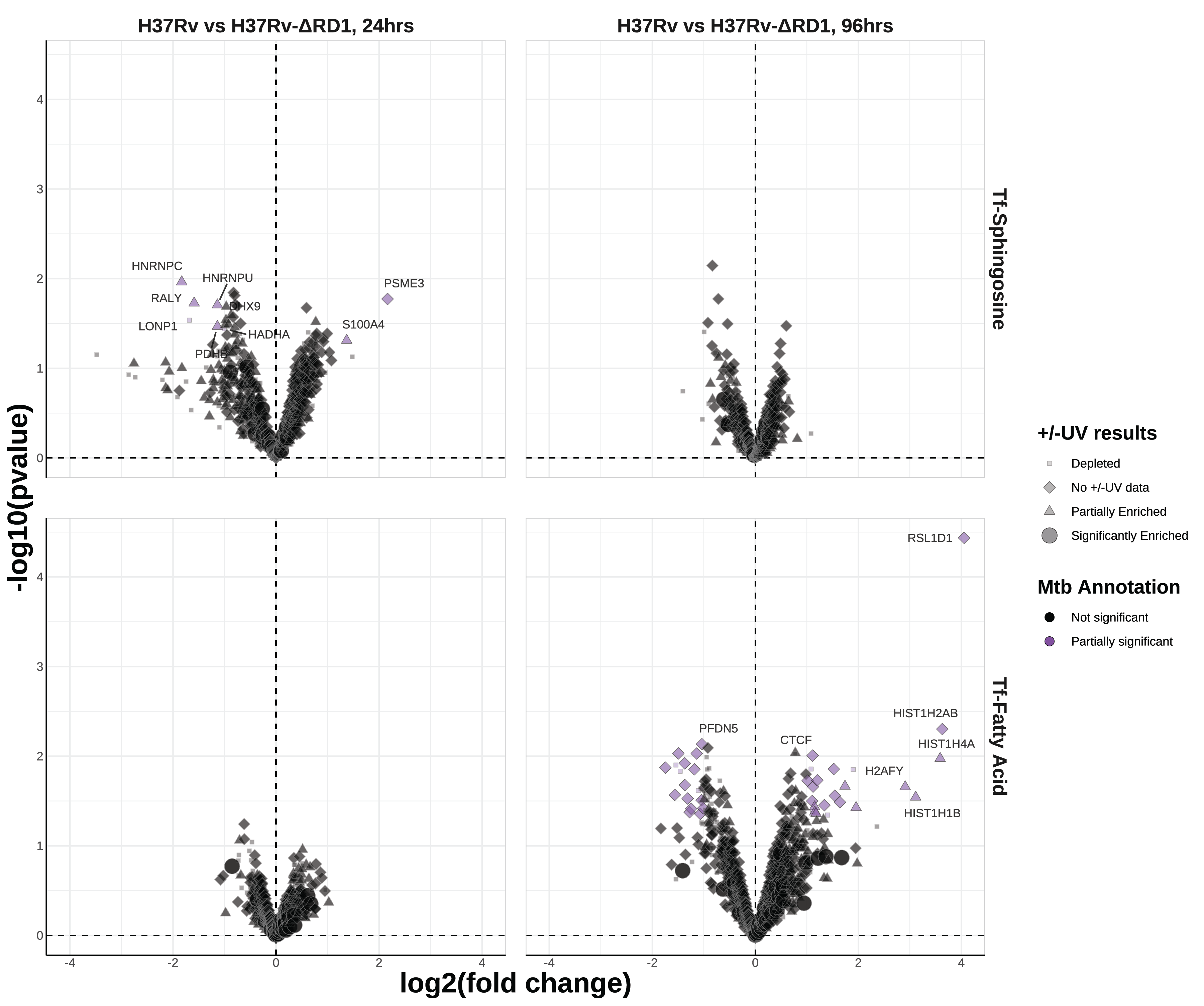
Despite this, we continued our assessment of H37Rv versus H37Rv-ΔRD1 by plotting the log2 fold-change of proteins during each infection. Again, very few proteins are substantively different between pathogenic H37Rv infection and attenuated H37Rv-ΔRD1 infection (Figure 6.6). Among these few proteins of partial significance, we see PSME3 enriched toward H37Rv infection at 24 hours; this protein is a component of the immunoproteasome critical for antigen presentation and correlated with antigen presentation31. At 96 hours post-infection, we see a mild enrichment of several Type I Interferon-regulating genes, including poly(ADP-ribose) polymerase family member 9 (PARP9), 2’-5’-Oligoadenylate Synthetase 3 (OAS3), and MX Dynamin Like GTPases 1 and 2 (MX1 and MX2). Interestingly, PARP9 is involved in protection against Mtb32, OAS3 restricts intracellular Mtb replication and enhances cytokine secretion33, and MX1 and MX2 are well known interferon-stimulated genes transcriptionally upregulated during Mtb infection24,34.
The only notable enriched gene for tf-8:3-FA was RSL1D1, enriched at 96 hours; this gene is known to suppress autophagy35. There is no reported connection to Mycobacterium tuberculosis.
6.5 Mtb protein Rv3466 is enriched by tf-Sphingosine during infection
The enrichment of mycobacterial proteins to either trifunctional sphingosine would be of profound significance – this would suggest direct evidence that mycobacteria utilize host sphingolipids during infection. The standard, though strict, criteria for confident protein identification above (at least two unique peptides identified in at least two replicates) yields no Mtb proteins. That we fail to identify Mtb proteins with high confidence may be expected in retrospect, given the disparity in total protein mass between an infected cell and an infecting bacillus. However, Mtb candidate peptides were identified aligning with the Mtb proteins accD2, Rv1910, and Rv3466 (Figure 6.7A). accD2 was only identified in the tf-8:3-FA TMT-16-plex, Rv3466 was only identified in the tf-Sph TMT-16-plex, and the same Rv1910c peptide was identified in both TMT-16-plexes. The relative enrichment of each protein toward H37Rv or H37Rv-ΔRD1 at both time points is depicted in Figure 6.7B. Among these three hits, only Rv3466 is enriched compared to mock infection for both strains at both time points.
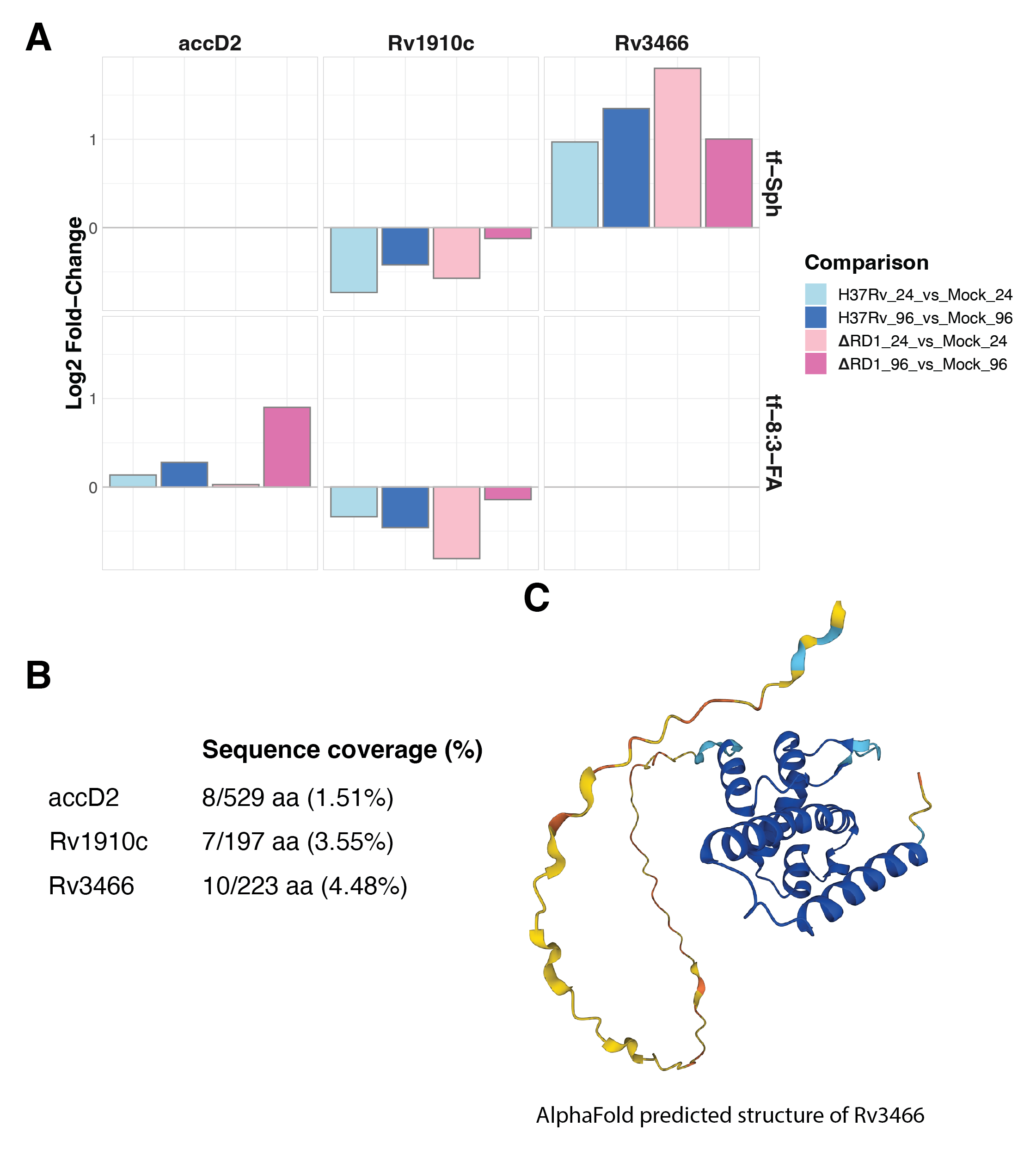
The total knowledge regarding Rv3466 can be summarized in a few words: it is an uncharacterized protein with no functional annotation. Benq et al. report that Rv3466 lies in an MT-complex-specific genomic island – the genes that distinguish environmental mycobacteria from pathogenic M. tuberculosis complex members36. A 2019 genome-wide transposon mutagenesis study by Minato et al. showed that this protein is not essential for in vitro growth in MtbYM-rich medium37; its expression is significantly enriched following culture with the broad-spectrum antibiotic levofloxacin38; no protein domains are annotated in the sequence of Rv3466. Rv3466 is enriched to the lipid fraction in studies of the total Mtb proteome39,40. This gene is close genomic neighbors with rmlB and rmlC, two sugar-modifying enzymes involved in synthesizing peptidoglycan – suggesting a role for Rv3466 in peptidoglycan synthesis at the bacterial membrane. Rv3466 shares high sequence homology with another uncharacterized Mtb protein, Rv1588c.
Further mass spectrometry analysis will determine whether these or other Mtb protein interactors of sphingosine can be identified.
6.6 Discussion
In this study, we expand the application of trifunctionalized probes to assess the impact of intracellular bacterial infection on the sphingolipid interactome. We first identified the proteins that selectively interact with trifunctionalized sphinganine and sphingosine (as opposed to a pair of control trifunctionalized fatty acid probes). We identify a suite of differentially enriched protein interactors of trifunctional sphingosine upon infection with the pathogenic Mtb strain H37Rv. Most differentially enriched proteins do not align with published transcriptional signatures of Mtb infection, suggesting that these proteins’ stability, localization, or function is altered during infection. We then compared the enriched interactions of cells infected with pathogenic and attenuated strains of Mtb. Finally, we hone in on a candidate Mtb protein enriched during infection (though its identification is below the confidence threshold).
Trifunctional probes are a precious resource for studying lipid biology. In this study, we primarily utilized these synthetic analogs of sphinganine and sphingosine as affinity handles to identify the protein interactors of these lipids in monocyte-derived macrophages. We note that a substantive majority of the enriched proteins either contain a transmembrane domain or are annotated as peripheral membrane proteins in the literature; very few are cytosolic or secreted. We identify several interactors reported in previous studies, such as Farley et al. 20246. Such previously verified hits include ECH1, CTSD, VDACs 1 and 2, PITPNB, PRCP, and PSAP. We identify several intriguing, novel binding partners that appear to be unique to macrophages – these include NENF, CSTA, SCPDH, and HSPA5, among others. More work is needed to validate these interactions and to determine the biological effects of these interactions.
Before we can discuss the results of the Mtb infection interactomics experiments, we must make a technical note about +/-UV proteomics: we would have expected a dramatic disparity between the total protein input of +UV samples and -UV samples due to the stringency of sample wash. Notably, there are proteins substantially enriched to the -UV sample condition for tf-Sphingosine and tf-Sphinganine, though we expect little biological significance among these proteins. These proteins are likely the result of spurious interactions, spontaneous crosslinking, or artifacts of data normalization. Notably, the depth of coverage in the +/-UV results hinders its utility in filtering out candidates – there were over 300 proteins identified in the Mtb infection samples treated with tf-Sph and 542 proteins identified in the samples treated with tf-8:3-FA. It is unclear how to consider these proteins which have no annotation in the baseline interactome.
With the baseline interaction analysis in mind, we may now discuss the main results of this study. We hoped to determine whether Mtb infection induces changes in the protein-sphingolipid interactions of the cell. We found that a 24-hour infection with H37Rv induces significant differential enrichment of 151 proteins (31 were enriched during infection, and 120 were depleted). Our comparison to publicly available transcriptomics data shows that while some transcriptionally upregulated proteins are reflected by positive interaction enrichment, little change is reported for many of the most trustworthy hits. While this comparison does not preclude changes in the total abundance of proteins during Mtb infection, it serves to decouple our interaction analysis from the transcriptional response of the cell.
There are many potentially exciting hits for follow-up. There appear to be several pathways enriched in the up- and downregulated interactions. These include an upregulation of immunity related proteins (e.g., MX1, IKBKB, and CTLA), cytoskeleton and motility proteins (e.g., myosin heavy and light chains, kinesin light chain, tubulin, and Coronin-7). Among the downregulated pathways are numerous secretory or vescicular trafficking proteins (e.g., COPE, GOLGA2, VAPA, and Rab11b). These results appear to suggest a significant dysregulation of cellular trafficking and endosomal protein composition. We also see a depletion of several calcium-binding proteins; this is intriguing because uncaging tf-sphingosine has been previously shown to induce the release of calcium from the lysosome9 – are these infected cells unable to trigger sphingosine-mediated calcium signaling? Finally, we report the low-confidence identification of a candidate Mtb protein selectively enriched to tf-Sphingosine during infection: Rv3466. This protein is not expected to contain a transmembrane domain and has no known function. Follow-up experiments are needed to validate this hit and to explore the possibility of further yet-undiscovered Mtb proteins that selectively interact with host sphingolipids.
While much work remains on this project, this study is among the first to utilize trifunctionalized lipids in an affinity enrichment of Mtb-infected cells. These mark crucial first steps into establishing a toehold into the waters of sphingolipid-mediated host-pathogen interactions. These results may yield new insights into the molecular mechanisms underlying the pathogenicity of Mycobacterium tuberculosis.
6.7 Materials and Methods
6.7.1 THP-1 cell culturing and plating
Human THP-1 monocytes were sourced from American Type Culture Collection (ATCC catalog no. TIB-22) were maintained in culture using RPMI 1640 medium (Thermo Fischer Scientific #11875119) supplemented with 10% fetal bovine serum (Seradigm), 1x Gibco penicillin-streptomycin (Thermo Fischer Scientific #15-140-163), and 1x Gibco Non-Essential Amino Acids (Thermo Fischer Scientific #11140050). Cells were grown in suspension at 37 °C with 5% CO2. Before experimental use, THP-1 monocytes were differentiated into macrophages via 48-hour treatment with 50 nM phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich P8139-1MG). All cell counting was achieved using a hemocytometer. For microscopy, cells were either plated in a 96-well plate at a density of 2.5 x 104 cells per well or on glass coverslips at 1.0 x 10 5 cells per well. For proteomics sample collection, cells were plated at a density of 4 x 106 cells per 10 cm dish.
6.7.2 Mycobacterium tuberculosis culturing and infection
Wildtype historical Mycobacterium tuberculosis strains H37Rv and H37Rv-ΔRD1 were gifts from the laboratory of Dr. David Lewinsohn at Oregon Health & Science University and Portland Veterans Association. Both Mtb strains were cultured in DifcoTM Middlebrook 7H9 medium (Beckton Dickinson, Cat# 271310) supplemented with BBLTM Middlebrook OADC Enrichment (Beckton Dickinson, Cat# L12240) and 0.1% Tween-80. Bacterial cultures were carefully monitored to guarantee they did not overgrow (OD600 > 1.5) and were kept at a low passage before experimental use.
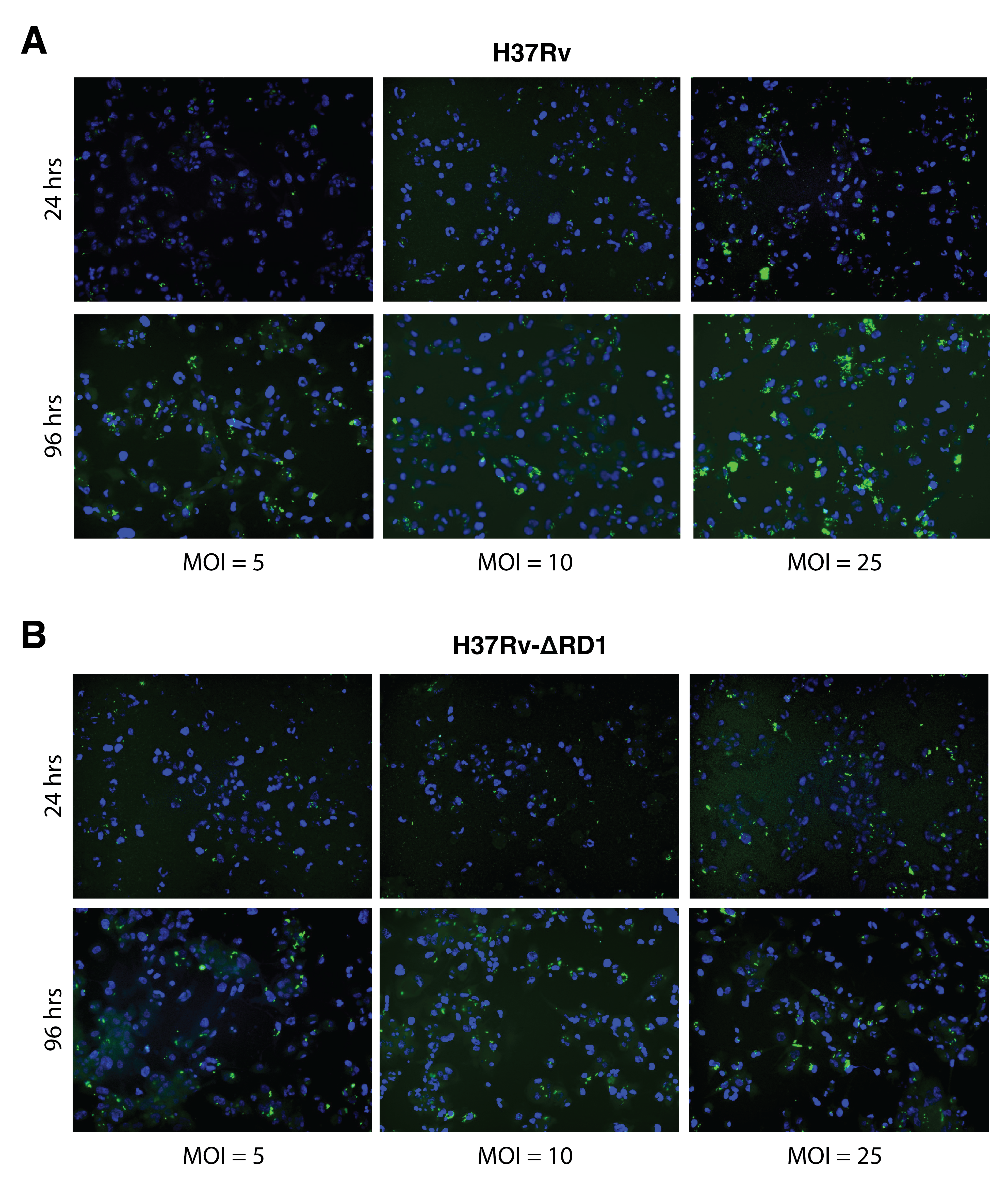
Before infection, THP-1 monocyte-derived macrophages were plated in 10 cm dishes at a density of 4 x 106 cells per dish and activated 48 hrs in 50 nM PMA. Mycobacterium tuberculosis (wildtype H37Rv and H37RvΔRD1) was cultured as above in 7H9 to mid-log phase and pelleted via 5 min spin at 2000 xg. Bacterial pellets were washed once with PBS and resuspended in RPMI. We prepared a single-cell suspension by passaging bacteria 25x through an 18-gauge blunt-tipped needle and then through a 5 μm filter. Bacterial concentration was estimated as a proportion of the OD600 (assuming 2.5 x 108 CFU/mL at an OD600 of 1.0). For 24-hour infection samples, cells were infected overnight at an MOI of 3. For 96-hour infection samples, cells were infected overnight at an MOI of 1 and washed 2x with PBS before an additional 72-hour incubation. Infections were repeated in 3 biological replicates on independent days. We validated a minimum infection rate of ~70% by plating parallel coverslips of activated THP-1 cells, which we infected with both strains of Mtb and at both MOIs and inspected via fluorescence microscopy.
6.7.3 Trifunctionalized lipid treatments, uncaging, crosslinking
After a 48-hour activation with 50 nM PMA, THP-1 cells were treated with a trifunctional lipid probe at the below concentrations in Table 6.2. Cells were incubated with probes for 30 min at 37 °C to allow probe incorporation into cellular membranes. Lipid probes were then exposed to blue wavelength light (405 nm) for 5 min to release the coumarin cage and incubated for the times depicted below (Table 6.2). Probes were then diazirine crosslinked via 5-minute exposure to ultraviolet wavelength light (355 nm). For proteomics experiments, control cells were treated with trifunctional probes and uncaged but were left without UV-induced diazirine crosslinking (-UV conditions)
6.7.4 Cell lysis and protein collection
After probe crosslinking, plates were washed 2x with ice-cold PBS, and cells were scraped into PBS using a cell scraper; for each sample, 2x 10 cm plates were combined during scraping for a total of 8 x 106 cells per sample. Cells were pelleted via 5 min spin at 500 xg and resuspended in a lysis buffer consisting of 8 M urea, 1x RIPA buffer (EMD Millipore, Cat# 20-188), and 1x HALT protease inhibitor cocktail (Thermo Fisher Scientific, Cat# 87786). Cell pellets were first broken up using repeated passage through an 18-gauge blunt-tipped needle, then passage through a lysis filter cartridge (from the MinuteTM Plasma Membrane Protein Isolation and Cell Fractionation Kit, Invent Biotech, Cat# SM-005) to achieve a solution of lysed cellular proteins and intact nuclei. All lysis steps were performed on ice or in a pre-cooled centrifuge.
Finally, in accommodation of OHSU biosafety level 3 protocol, cell lysates from Mtb-infected samples (and corresponding mock-infected controls) were spun through a 0.5 μm centrifugal filter (Ultrafree-MC GV Centrifugal Filter, EMD Millipore, Cat# UFC30GV25) via 20-minute spin at 10,000 xg.
6.7.5 Fluorescence click reaction & In-gel/on-blot fluorescence
In-gel fluorescence was used as a quality control metric for infected samples. 5% of each sample was split off and conjugated with CalFluor647-azide via CuAAC. 50 μL of each sample was incubated with 100 μM tris(benzyltriazolylmethyl)amine (TBTA), 1 mM cupric sulfate (CuSO4), 1 mM sodium ascorbate, and 20 μM CalFluor 647-azide (Click Chemistry Tools, Cat. #1372-1). Click reaction mixtures were incubated at room temperature for 60 minutes in the dark.
Following the click reaction, Laemmli sample buffer containing β-mercaptoethanol was added, and samples were heated to 42 °C for 30 minutes. Samples were spun at 10000 xg for 10 minutes. Each sample was loaded and was run on a 12.5% SDS-PAGE gel until the dye front had wholly run off the bottom of the gel – this removes much of the free, non-clicked 647-azide dye in each sample. After running, gels were left for at least 30 minutes in diH2O to allow any remaining free dye to diffuse out of the gel. Gels were then imaged using the 647nm filter of a Sapphire™ FL biomolecular fluorescence imager (Azure Biosystems). Contrast and brightness were globally adjusted in FIJI.
For validation of hits using on-blot fluorescence, samples were fluorescently labeled and separated via SDS-PAGE as above, and gels were transferred to polyvinylidene difluoride blotting membranes via overnight wet transfer. Blots were blocked using 1% BSA and stained with denoted primary antibodies. Blots were then stained against the appropriate primary background with a secondary antibody conjugated to Alexa Fluor 555 fluorophore. Finally, blots were imaged as above on a Sapphire™ FL biomolecular fluorescence imager using the 555 nm and 647 nm filters. Contrast and brightness were globally adjusted in FIJI.
6.7.6 Affinity purification of probe-bound proteins
The remaining 95% of each sample was used as input for affinity purification via CuAAC to azide beads. First, Picolyl Azide Agarose resin (Click Chemistry Tools, Cat. #1408-2; 50% slurry) was transferred to new Eppendorf tubes and gently spun to pellet; storage solution supernatant was aspirated and beads were washed with diH2O to remove any residual storage solution. CuAAC/click reaction was then initiated through the addition of 100 μM TBTA, 1 mM sodium ascorbate, 1 mM CuSO4, washed picolyl-azide agarose resin, and the remaining volume of probe-treated lysates. The final 1.25 mL reaction proceeded under vigorous shaking for 1 hour at room temperature.
6.7.6.1 Bead wash & digestion procedure
Bead/Click reaction mix transferred to a 2 mL gravity flow column (Pierce™ Centrifuge Columns 2 mL, Cat# 89896). Flow through was collected for each stage of the washing. Beads were washed a total of 25x with the buffers denoted below in Table 6.3.
| Composition | # of washes (volume per wash) | |
|---|---|---|
| Wash Buffer #1 | 100 mM tris-HCl, 250 mM NaCl, 5 mM ethylenediaminetetraacetic acid (EDTA), 1% sodium dodecyl sulfate (SDS) (pH = 8.0) | 5 (1 mL) |
| Wash Buffer #2 | 100 mM tris-HCl, 8 M urea (pH = 8.0) | 10 (2 mL) |
| Wash Buffer #3 | 20 % acetonitrile, 80 % diH2O (volume/volume) | 10 (2 mL) |
Beads were transferred to a new Eppendorf tube and pelleted. Bead-conjugated proteins were reduced via treatment with 1 mM dithiothreitol for 30 minutes at 42 °C. Beads were again pelleted and resuspended in an alkylating solution of 40 mM iodoacetamide for 30 minutes at room temperature (in the dark). Beads were then pelleted, and the alkylating solution was aspirated.
Finally, the beads were resuspended in 40 μL of a tryptic digestion solution consisting of 100 mM tris-HCl, 2 mM CaCl2, and 10% acetonitrile. A total of 1 μg of trypsin (Promega Trypsin/Lys-C Mix, Mass Spec Grade, Cat. #V5071) was added to each sample for a 1:25 ratio of [LC-MS-grade trypsin/Lys C : substrate] (assuming 1mg/mL protein on beads). Beads were rocked vigorously at 37 °C overnight to digest bead-conjugated proteins.
6.7.6.2 Sample desalting
Following digestion from beads, the digestion solution was washed and desalted using a reverse phase C18 desalting spin column (BioPureSPN MIDI, PROTO 300 C18, Fisher Scientific Cat #NC1678004). At all stages of desalting, columns were spun at 110 xg for 1 minute. First, columns were conditioned with acetonitrile (ACN) and flushed with H2O; columns were equilibrated with 2% ACN and blotted dry. The bead solution (including beads) was then transferred to the spin column and washed 2x with 2 % ACN to remove trace salts. The sample was released twice with an elution buffer of 80% ACN and 0.1 % trifluoroacetic acid. Finally, the eluted peptide solution was dried using a speedvac and stored at -80 °C.
6.7.7 Liquid chromatography and tandem mass spectrometry analysis (LC-MS/MS)
Mass spectrometric analyses were performed as previously described in Farley et al. 20246 and performed at the European Molecular Biology Laboratories Proteomics Core facility. Dried peptides were shipped to EMBL, labeled with isobaric mass tags using the TMT-16-plex system, and analyzed by LC-MS/MS on an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific).
Peptides were separated by reverse phase chromatography on an Ultimate 3000 nano RLSC system (Dionex) through a trapping cartridge (Precolumn C18 PepMap100, 5 mm, 300 μm i.d., 5 μm, 100 Å) and an analytical column (Acclaim PepMap 100, 75 x 50 cm C18, 3mm, 100 Å) connected to a nanospray-Flex ion source. The peptides were loaded onto the trap column at 30 μL/min using Solvent A (0.1% formic acid in H2O) and eluted using a gradient from 2 to 80% Solvent B (0.1% formic acid in acetonitrile) over 2 hr at 0.3 μL/min (all solvents were LC-MS grade). The Orbitrap Fusion Lumos was operated in positive ion mode with a spray voltage of 2.2 kV and a capillary temperature of 275 °C. Full scan MS spectra with a mass range of 375-1500 m/z were acquired in profile mode using a resolution of 120,000 with a maximum injection time of 50 ms, AGC operated in standard mode, and an RF lens setting of 30%. Fragmentation was triggered for 3 s cycle time for peptide-like features with a charge state of 2-7 on the MS scan (data-dependent acquisition).
Fragmentation was triggered for 3 second cycle time for peptide-like features with charge states of 2-7 on the MS scan (in data dependent acquisition). Precursors were isolated using the quadrupole with a window of 0.7 m/z and fragmented with a normalized collision energy of 34%. Fragment mass spectra were acquired in profile mode and with a resolution of 30,000. The maximum injection time was set to 94 ms and AGC target set to custom. The dynamic exclusion was set to 60 s6.
6.7.8 Data analysis
Dr. Frank Stein prepared the data analysis pipeline at the European Molecular Biosciences Laboratories in Heidelberg, Germany and performed the analysis as previously reported in Farley et al., 20246. The acquired MS data were analyzed using IsobarQuant41 and Mascot V.24 (Matrix Science) using a reverse UniProt FASTA database including common contaminants; Mtb infection data were analyzed using a concatenated UniProt FASTA database including both human and Mycobacterium tuberculosis protein sequences. Only proteins with at least two unique peptides with lenght of at least 7 amino acids and a false-discovery rate below 0.01 were kept for protein-level analysis. The acquired spectra were scanned for the following peptide modifications: Carbamidomethyl (C, fixed), TMT-16-plex (K, fixed), Acetyl (N-term, variable), Oxidation (M, variable), TMT-16-plex (N-term, variable). The mass error tolerance for full-scan MS spectra was set to 10 ppm and for MS/MS spectra to 0.02 Da. A maximum of 2 missed cleavage sites was allowed. Batch effects were removed between replicates, a variance stabilization normalization was applied on the log2-transformed raw data42, and changes in abundance of proteins in the experimental condition versus control condition were calculated using LIMMA analysis14.
For the +/-UV experiment of baseline protein-lipid probe interactions, a protein was designated “significantly enriched” if it was enriched toward the +UV condition by at least a log2 fold-change of 1 and a p-value < 0.05. Proteins enriched toward the +UV condition but with p-value > 0.05 were designated “partially enriched”. Due to the larger number of differentially enriched proteins in the Mtb infection affinity proteomics experiment, we used a modified designation of significance: proteins were deemed “significant” if they had a log2 fold-change enrichment of magnitude > 2 (both positive and negative), a p-value < 0.01 and a false-discovery rate < 0.05; proteins were deemed “partially significant” if the magnitude of their log2 fold-change enrichment was greater than 1 (in either the positive or negative direction) and a p-value < 0.05.
Data visualization was performed using R. Packages included: Bioconductor, ggplot2, dplyr, vsn, limma, MSnbase, gplots, fdrtool, biobroom, tidyverse, ggrepel.
Comparative transcriptomic data was found using the GEO database at the National Center for Biotechnology Information, initially reported by Jani et al. in 202324.
6.8 Contributions
GG designed the study, performed sample collection and analyzed results, prepared visualizations, prepared the figures, and wrote the manuscript. SF assisted in designing mass spectrometry experiments, designed and synthesized lipid probes, assisted in data visualization, and edited the manuscript. PH & FS assisted in designing mass spectrometry experiments, performed mass spectrometry and protein identification, and assisted in the interpretation of results. CS assisted in designing mass spectrometry experiments, oversaw the design and synthesis of lipid probes, provided reagents, and gave expert advice on study design. FT assisted in the study design and oversaw the study.
6.9 Supplemental Data